
- Detalles
- Categoría: BIOVIA
- Visto: 545
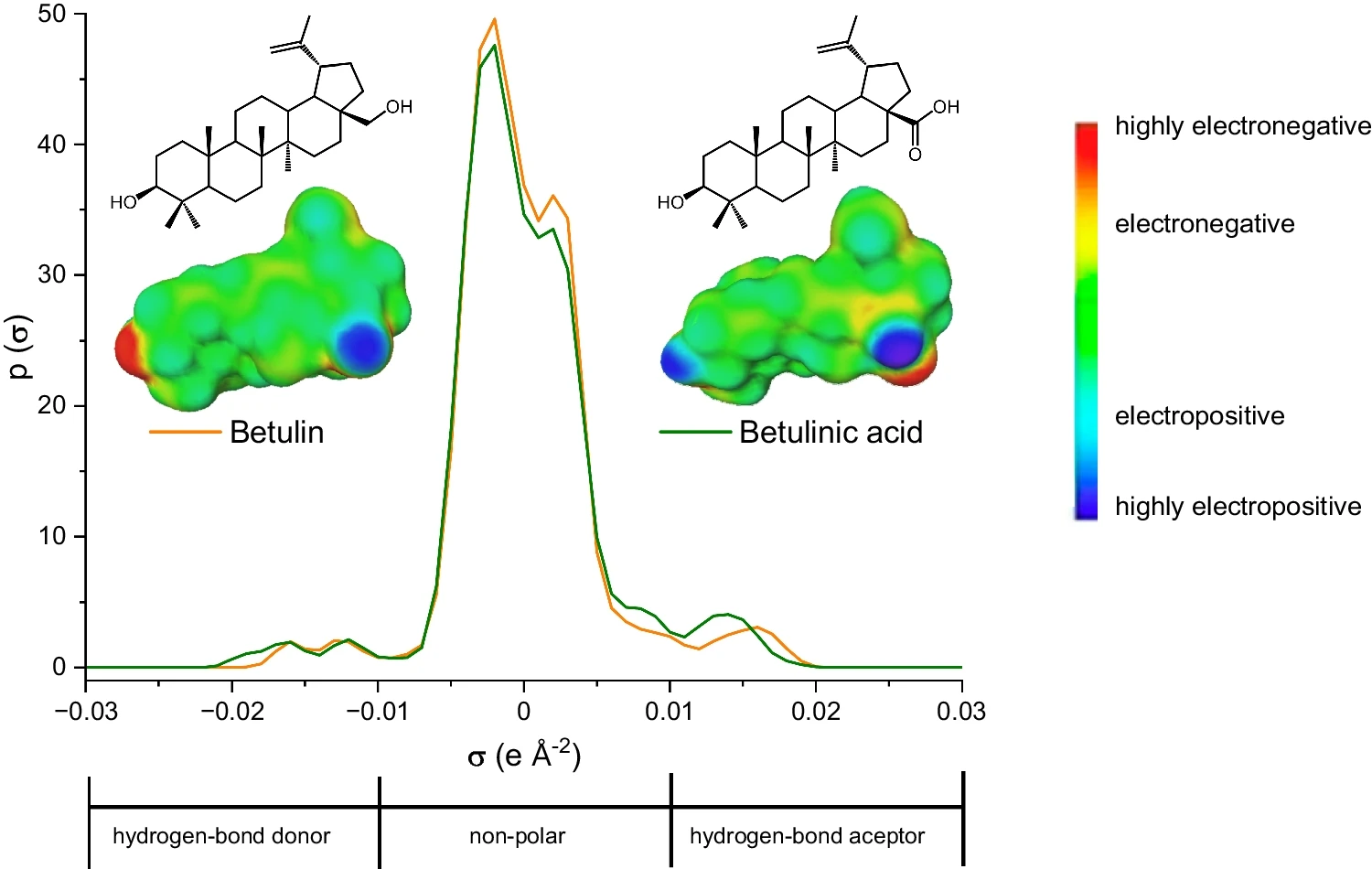 Figura 1. Estructuras químicas, superficies sigma y perfiles sigma de betulina y ácido betulínico estimados mediante COSMO-RS.
Figura 1. Estructuras químicas, superficies sigma y perfiles sigma de betulina y ácido betulínico estimados mediante COSMO-RS.Este trabajo [1] ha sido desarrollado por el…
- Detalles
- Categoría: BIOVIA
- Visto: 747
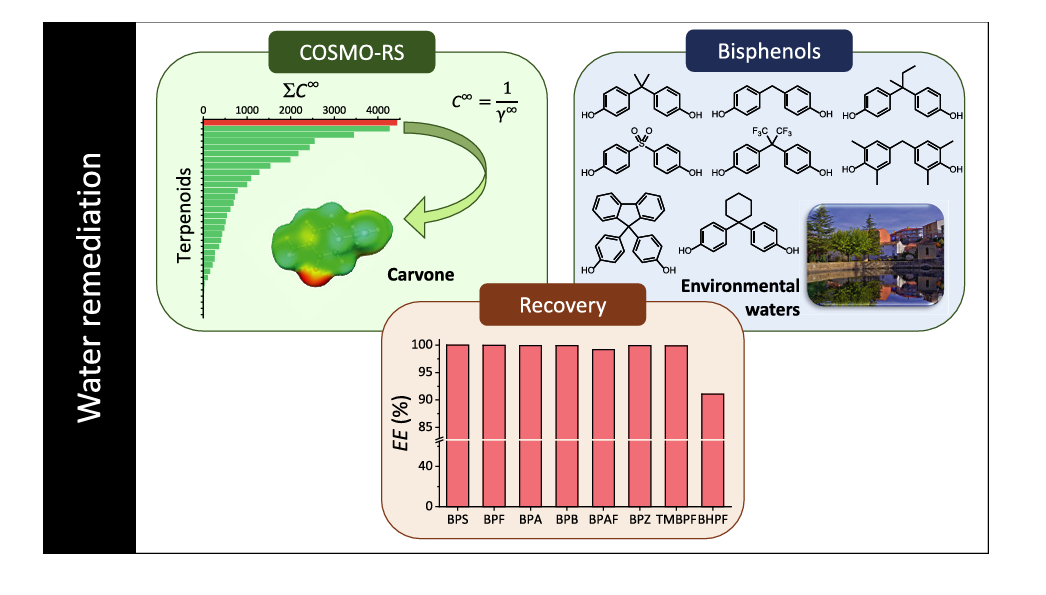
Este trabajo [1] ha sido desarrollado por el equipo investigador formado por Luz Alonso Dasques, Plácido Galindo Iranzo, Rosa Lebrón Aguilar y Jesús E. Quintanilla López, pertenecientes al Instituto de Química-Física “Blas Cabrera” (IQF-CSIC), e Iván Sacristán y Belén Gómara, pertenecientes al Instituto de Química…
- Detalles
- Categoría: BIOVIA
- Visto: 722
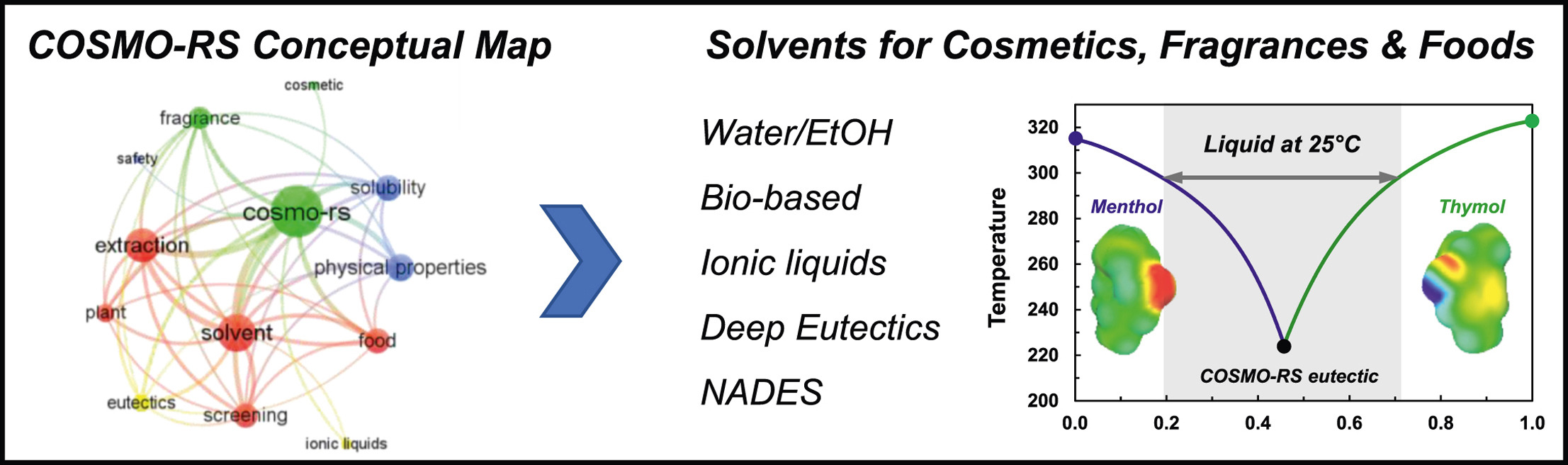
Un reciente artículo publicado en Current Opinion in Colloid & Interface Science analiza en detalle esta evolución, destacando el crecimiento del uso de COSMO-RS en formulación y su papel clave en el desarrollo de soluciones más sostenibles. El trabajo revisa aplicaciones en alimentación y otros sectores como la cosmética y las fragancias, mostrando cómo estas herramientas están ganando protagonismo en entornos industriales. Si quieres profundizar en esta tendencia y ver ejemplos concretos, puedes consultar el estudio completo.
Una…
- Detalles
- Categoría: BIOVIA
- Visto: 1473
Cálculos más robustos y confiables
TURBOMOLE 2026 incluye parámetros por defecto optimizados que permiten iniciar simulaciones con configuraciones más adecuadas, reduciendo errores y asegurando resultados consistentes en química cuántica.
- Mayor estabilidad en cálculos DFT y COSMO
- Menos ajustes manuales requeridos
- Workflows más ágiles para investigadores