
Modelado de polímeros de precisión: aprovechamiento de las innovaciones de Materials Studio y Scripting
- Detalles
- Categoría: BIOVIA
- Visto: 2122
Materials Studio ofrece una plataforma fácil de usar pero potente para modelar una amplia gama de sistemas, y este informe se centra específicamente en polímeros y redes de polímeros. Junto con Pipeline Pilot, hay varios métodos disponibles para modelar materiales tan complejos que van desde el polietileno reticulado (XLPE) ampliamente reportado, hasta redes personalizadas con mecanismos de reacción únicos. Los campos de fuerza atómicos unidos se desarrollan para modelar de manera eficiente tales sistemas que involucran grandes cantidades de partículas, proporcionando un equilibrio entre la eficiencia computacional y la precisión.
Los campos de fuerza son fundamentales en la simulación molecular, ya que determinan cómo interactúan las partículas a través de enlaces y otras interacciones. Estos campos son cruciales para predecir propiedades macroscópicas de sistemas como polímeros, incluyendo densidad y temperatura de transición vítrea. Existen varios campos de fuerza, como COMPASSIII y OPLS, cada uno optimizado para diferentes materiales. Además, se utilizan métodos de átomo unido y grano grueso para simplificar simulaciones, permitiendo pasos de tiempo más largos y una mayor eficiencia computacional.
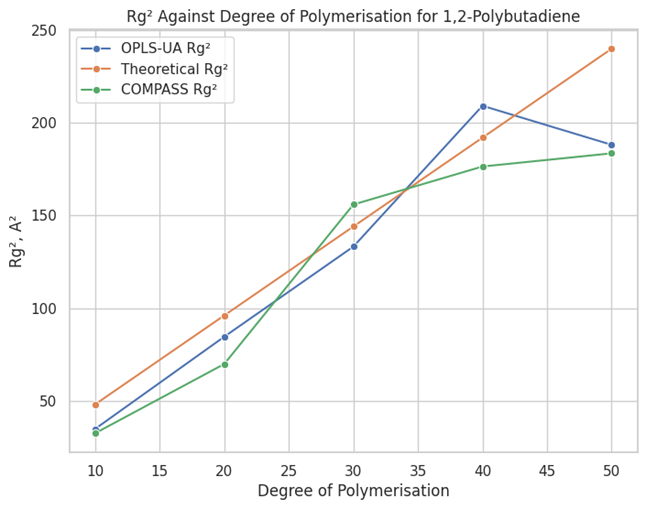
En Materials Studio, se desarrolló un campo de fuerza personalizado OPLS-UA mediante la modificación de parámetros atomísticos y validación de torsiones a través de análisis conformacional. Se integraron secuencias de comandos en Perl para extraer energía de torsión y se compararon resultados con la literatura. El campo se probó en seis sistemas de polímeros, equilibrándolos mediante simulaciones de dinámica molecular. Para automatizar el proceso de cálculo del radio de giro, se crearon protocolos Pipeline Pilot, lo que redujo el tiempo y el error humano, validando así la precisión del campo de fuerza.
Se desarrolló un método para convertir polímeros de una representación atomística completa a una de átomo unido eliminando hidrógenos y ajustando los tipos de campo de fuerza. Se creó un script en Perl para automatizar este proceso. Con el campo de fuerza OPLS-UA validado, se pueden modelar reacciones como adición y cicloadición en Materials Studio. Se utilizó el polietileno reticulado (XLPE) como ejemplo, donde se modeló su red mediante un protocolo Pipeline Pilot, ajustando las probabilidades de reacción para investigar sus efectos en las propiedades físicas.
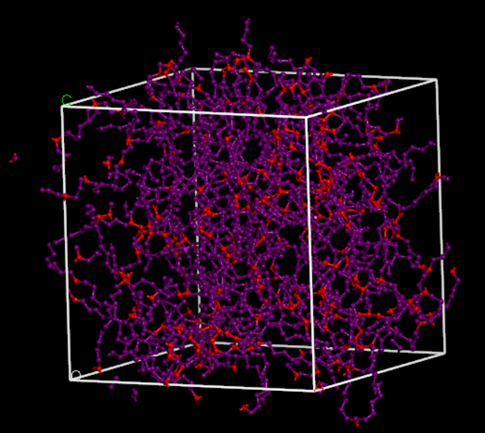 Fragmento de XLPE que muestra los enlaces cruzados formados entre cadenas. Los átomos están coloreados por tipo de campo de fuerza.
Fragmento de XLPE que muestra los enlaces cruzados formados entre cadenas. Los átomos están coloreados por tipo de campo de fuerza.
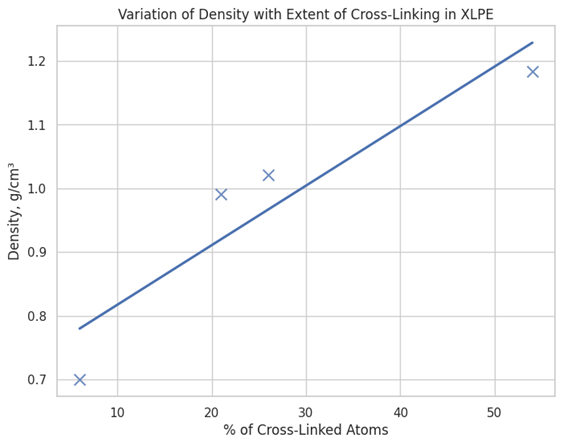 Efecto del grado de reticulación en modelos XLPE equilibrados sobre la densidad.
Efecto del grado de reticulación en modelos XLPE equilibrados sobre la densidad.
Las reacciones de Diels-Alder son importantes en la cicloadición de un dieno con un alqueno, permitiendo la unión de monómeros en sistemas poliméricos complejos. La herramienta Buscador de reacciones se utiliza para dibujar reactivos y productos, mapeando átomos y identificando contactos cercanos entre ellos, lo que proporciona un control significativo. Esto es especialmente útil para modelar estructuras específicas que presentan sitios reactivos atípicos.
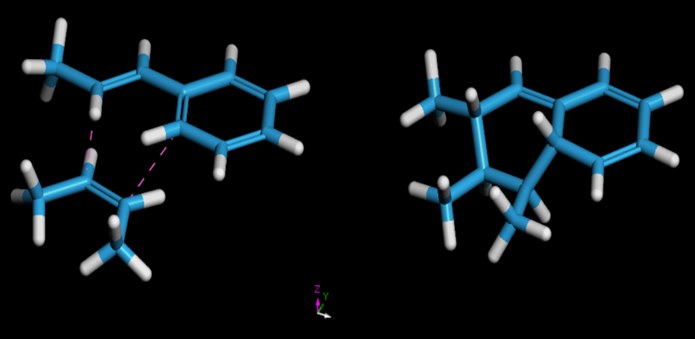 Reactivos (izquierda) con contactos cercanos definidos para una reacción de Diels-Alder para formar productos (derecha).
Reactivos (izquierda) con contactos cercanos definidos para una reacción de Diels-Alder para formar productos (derecha).
Para modelar reacciones de condensación, se utilizó un script Perl personalizable que define los átomos reactivos en monómeros y agentes de curado, organizándolos en una célula amorfa. Este script especifica los átomos reactivos junto con la estructura de entrada y utiliza una subrutina para definir eventos de ruptura de enlaces, produciendo el producto deseado. Su versatilidad permite la reticulación de diversos monómeros y agentes, adaptándose a diferentes requisitos del sistema.
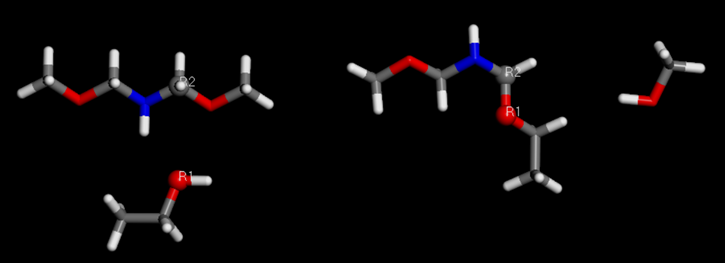 Reactivos (izquierda) con átomos reactivos R1 y R2 definidos para formar el producto (derecha).
Reactivos (izquierda) con átomos reactivos R1 y R2 definidos para formar el producto (derecha).
Materials Studio es una plataforma destacada para modelar sistemas y redes de polímeros complejos, ofreciendo diversas herramientas y opciones de personalización. Su integración con Pipeline Pilot, el desarrollo de campos de fuerza personalizados y las secuencias de comandos de Perl brindan un control preciso en la construcción de modelos y simulaciones. Esta versatilidad y control permiten a los investigadores avanzar en el diseño, optimización y predicción de propiedades de polímeros con alta precisión y eficiencia.
La importancia de los campos de fuerza en la simulación molecular
Los campos de fuerza son fundamentales en la simulación molecular, ya que determinan cómo interactúan las partículas a través de enlaces y otras interacciones. Estos campos son cruciales para predecir propiedades macroscópicas de sistemas como polímeros, incluyendo densidad y temperatura de transición vítrea. Existen varios campos de fuerza, como COMPASSIII y OPLS, cada uno optimizado para diferentes materiales. Además, se utilizan métodos de átomo unido y grano grueso para simplificar simulaciones, permitiendo pasos de tiempo más largos y una mayor eficiencia computacional.
Desarrollo de campo de fuerza personalizado en Materials Studio
En Materials Studio, se desarrolló un campo de fuerza personalizado OPLS-UA mediante la modificación de parámetros atomísticos y validación de torsiones a través de análisis conformacional. Se integraron secuencias de comandos en Perl para extraer energía de torsión y se compararon resultados con la literatura. El campo se probó en seis sistemas de polímeros, equilibrándolos mediante simulaciones de dinámica molecular. Para automatizar el proceso de cálculo del radio de giro, se crearon protocolos Pipeline Pilot, lo que redujo el tiempo y el error humano, validando así la precisión del campo de fuerza.
Se desarrolló un método para convertir polímeros de una representación atomística completa a una de átomo unido eliminando hidrógenos y ajustando los tipos de campo de fuerza. Se creó un script en Perl para automatizar este proceso. Con el campo de fuerza OPLS-UA validado, se pueden modelar reacciones como adición y cicloadición en Materials Studio. Se utilizó el polietileno reticulado (XLPE) como ejemplo, donde se modeló su red mediante un protocolo Pipeline Pilot, ajustando las probabilidades de reacción para investigar sus efectos en las propiedades físicas.
Fragmento de XLPE que muestra los enlaces cruzados formados entre cadenas. Los átomos están coloreados por tipo de campo de fuerza.Efecto del grado de reticulación en modelos XLPE equilibrados sobre la densidad.Las reacciones de Diels-Alder son importantes en la cicloadición de un dieno con un alqueno, permitiendo la unión de monómeros en sistemas poliméricos complejos. La herramienta Buscador de reacciones se utiliza para dibujar reactivos y productos, mapeando átomos y identificando contactos cercanos entre ellos, lo que proporciona un control significativo. Esto es especialmente útil para modelar estructuras específicas que presentan sitios reactivos atípicos.
Reactivos (izquierda) con contactos cercanos definidos para una reacción de Diels-Alder para formar productos (derecha).Para modelar reacciones de condensación, se utilizó un script Perl personalizable que define los átomos reactivos en monómeros y agentes de curado, organizándolos en una célula amorfa. Este script especifica los átomos reactivos junto con la estructura de entrada y utiliza una subrutina para definir eventos de ruptura de enlaces, produciendo el producto deseado. Su versatilidad permite la reticulación de diversos monómeros y agentes, adaptándose a diferentes requisitos del sistema.
Reactivos (izquierda) con átomos reactivos R1 y R2 definidos para formar el producto (derecha).Conclusión - El Poder de los Materiales Estudio para el Modelado de Redes de Polímeros utilizando Campos de Fuerza de Átomos Unidos
Materials Studio es una plataforma destacada para modelar sistemas y redes de polímeros complejos, ofreciendo diversas herramientas y opciones de personalización. Su integración con Pipeline Pilot, el desarrollo de campos de fuerza personalizados y las secuencias de comandos de Perl brindan un control preciso en la construcción de modelos y simulaciones. Esta versatilidad y control permiten a los investigadores avanzar en el diseño, optimización y predicción de propiedades de polímeros con alta precisión y eficiencia.