
Predicción de espectros RMN en ChemDraw y Chem3D
- Detalles
- Categoría: ChemOffice
- Visto: 44040
ChemDraw y Chem3D tienen herramientas para predecir espectros de RMN e IR. Esta noticia discutirá las características y métodos clave para esta predicción.
Algunos paquetes de química computacional incluyen predicciones de RMN y otros, sin embargo, predicen IR. Uno de los paquetes de química computacional, GAMESS, está integrado en Chem3D e incluido, por tanto, en el paquete de ChemOffice.
ChemNMR: predicción de RMN en ChemDraw
Este paquete está completamente integrado con ChemDraw, el usuario puede dibujar una molécula y obtener de forma sencilla su espectro RMN. Para poder visualizar las predicciones 1H-NMR y 13C-NMR se debe seleccionar la molécula objetivo y, a continuación, seleccionar el menú “estructura – predecir 1H-NMR” o “13C-NMR”. ChemDraw mostrará la información y el espectro en una nueva ventana manteniendo la molécula original sin cambios en la ventana original.
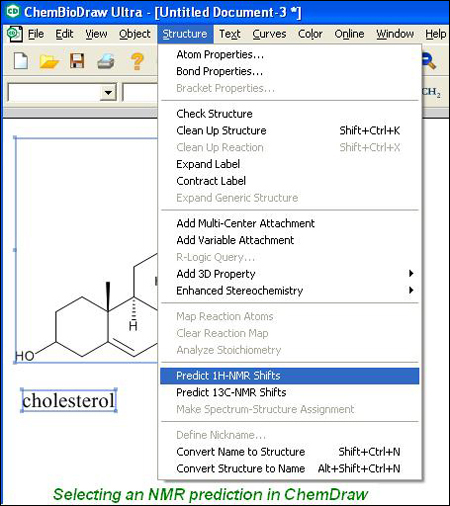
La nueva ventana presentará varias características a tener en cuenta:
- Cada hidrógeno está marcado con un número que indica su desplazamiento en ppm.
- Colocando el cursor sobre cualquier hidrógeno (o hidrógeno implícito) de la molécula, aparecerán marcadas en verde las señales correspondientes a ese núcleo.
- De igual manera, al colocar el cursor sobre cualquier señal del espectro aparecerá marcado el hidrógeno correspondiente en la molécula a estudiar.
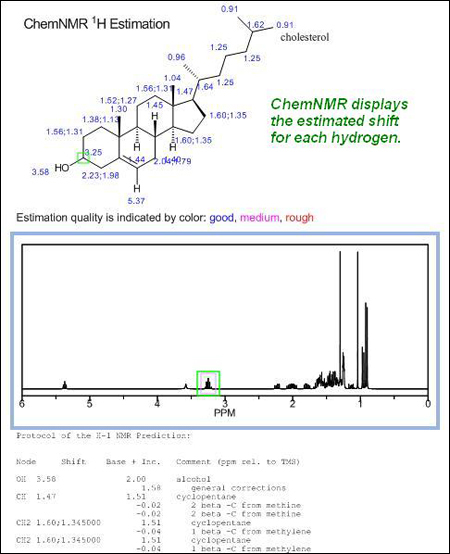
Configuración de parámetros de NMR en ChemDraw
A menudo los usuarios de ChemDraw se preguntan si existe la posibilidad de seleccionar el disolvente en que se realizará el espectro NMR, ChemNMR no puede hacer eso. En cambio, en las predicciones que se realicen en Chem3D si podrá seleccionarse el disolvente y muchos otros parámetros.
Indicar además que la forma de la molécula en ChemDraw es irrelevante a la hora de predecir espectos, pues ChemNMR utilizará la versión “limpia” de la molécula.
Hay un parámetro que si puede modificarse en ChemNMR, aunque solo para 1H-NMR, la intensidad de campo. Por defecto, la intensidad de campo se establece en una frecuencia de espectrómetro de 300 MHz. Los espectros mostrados anteriormente utilizaban esa intensidad de campo. Para cambiar la intensidad se deberán seguir los siguientes pasos:
Seleccione la molécula con cualquiera de las herramientas de selección de ChemDraw y pulse “Alt”, mantenga pulsada esta tecla y seleccione mientras “estructura – predicción 1H-NMR”, en lugar de producirse el espectro de resonancia, aparecerá un cuadro de diálogo que le indicará con qué frecuencia se va a realizar el espectro y podrá modificarla.
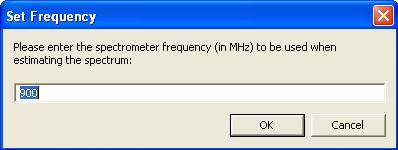
Tenga en cuenta que la intensidad que establezca permanecerá como valor establecido hasta que vuelva a cambiarlo, incluso después de salir de ChemDraw.
Una vez modifique el valor y presione “ok” podrá volver a seleccionar “estructura – predicción 1H-NMR” para que ChemDraw le ofrezca el espectro con la intensidad de campo seleccionada. En esta ocasión no es necesario mantener presionada la tecla “Alt”.
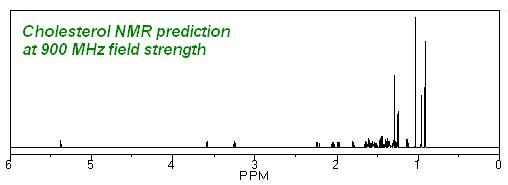
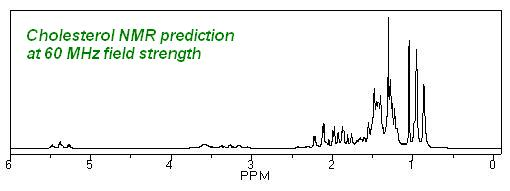
GAMESS: Predicción de NMR e IR en Chem3D
El paquete de química computacional GAMESS incluye predicción RMN e IR. A partir de este paquete computacional se pueden obtener espectros para moléculas que ChemNMR consideraría iguales. Se realizará el estudio de la molécula 7,7-dimetilnorborneno, que contiene dos grupos metilo que ChemDraw considera equivalentes pero que, en cambio, Chem3D diferencia que no lo son.
La estructura Chem3D aclara la diferencia entre los dos grupos metilo: uno está cerca de un doble enlace en el anillo y el otro está cerca de un enlace simple. La siguiente estructura de Chem3D es la estructura creada por la interfaz por defecto, sin minimización de energía, por lo tanto, es solo un cálculo geométrico basado en evitar obstáculos estéricos y otras reglas estándar. Por lo tanto, vemos una ventaja inherente en observar moléculas en 3D sobre 2D.
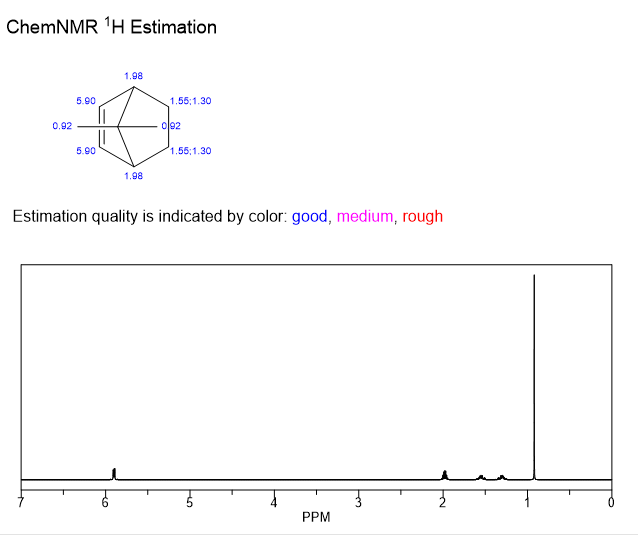
A continuación se realiza una predicción inicial a partir de ChemNMR en ChemDraw para poder llevar a cabo una comparación. ChemNMR asigna a los dos grupos metilo la misma predicción de desplazamiento (0,92 ppm) porque los considera químicamente equivalentes.
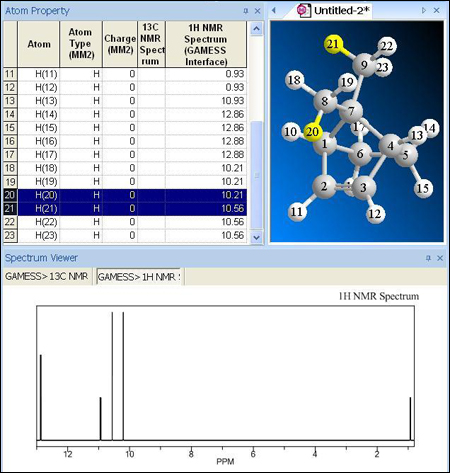
Al observar la misma predicción de RMN a través de GAMESS se observan diferencias significativas. GAMESS indica que el desplazamiento para el grupo metilo que se encuentra cerca del doble enlace será 10,21 ppm mientras que el metilo que se encuentra cerca del enlace simple presenta un desplazamiento de 10,56 ppm. En la siguiente imagen el hidrógeno 21 pertenece al metilo cerca del enlace simple y el hidrógeno 20 pertenece al metilo cerca del doble enlace. Hay que tener en cuenta que GAMESS no predice el acoplamiento entre núcleos, en cambio ChemNMR si lo hace.
Debemos tener en cuenta que ChemDraw y Chem3D proporcionan información RMN e IR para múltiples compuestos. Cada método tiene sus propias fortalezas, pero en conjunto, satisfacen las necesidades del investigador. ChemDraw ofrece el acoplamiento entre núcleos de protón en RMN mientras que GAMESS ofrece la posibilidad de utilizar diferentes disolventes y distinguir entre núcleos aparentemente idénticos, pero que por entorno no lo son.
- Está aquí:
-
Inicio

-
Noticias
-
ChemOffice
- Predicción de espectros RMN en ChemDraw y Chem3D