
De la teoría a la práctica: aprovechamiento de COSMO-RS para predicciones de pKa ácido en acetona
- Detalles
- Categoría: BIOVIA
- Visto: 3544

Explorando la química cuántica
El pKa de un ácido es una medida de su fuerza, que es un factor crucial en su reactividad. La predicción precisa de los valores de pKa nos permite comprender la reactividad relativa de diferentes ácidos, lo que puede ser importante en varios procesos químicos, como la síntesis, la catálisis y la bioquímica. También puede guiar el desarrollo de nuevos materiales con las propiedades deseadas. Veamos qué tan bien se desempeña la química cuántica en la evaluación de esta propiedad esencial examinando los resultados de un estudio conjunto realizado por investigadores de la Universidad del Ruhr de Bochum y BIOVIA [1].El disolvente investigado es la acetona. La acetona es un material versátil con una amplia gama de aplicaciones. En la vida cotidiana, es quizás mejor conocido por sus capacidades de limpieza como quitaesmalte. Sin embargo, también es un disolvente esencial en diversas industrias. La acetona se utiliza como disolvente para extraer compuestos de muestras para su análisis, para disolver resinas y recubrimientos o ingredientes farmacéuticos activos (API) y otros componentes de formulaciones farmacéuticas, y para desinfectar y limpiar instrumentos. Es un disolvente común en química orgánica, que permite una amplia gama de reacciones.
La metodología computacional de elección es COSMO-RS, también conocida como Modelo de Cribado Similar a Conductor para Disolventes Reales. Es una poderosa herramienta computacional con amplias aplicaciones en diversas industrias. Su capacidad para predecir e interpretar diversas propiedades fisicoquímicas de compuestos en diferentes disolventes lo convierte en un activo valioso en muchas industrias, por ejemplo, la ingeniería química, la ciencia de los materiales, la farmacéutica, la agroquímica y las industrias de sabores y fragancias. COSMO-RS se puede aplicar para diseñar nuevos productos, optimizar procesos y garantizar el cumplimiento de las normativas medioambientales.
Explorando el vínculo entre el pKa y las predicciones cuánticas para obtener información sobre las reacciones
Se puede aplicar una relación lineal de energía libre (LFER) entre el valor de pKa y el cambio de la energía libre de disociación de Gibbs (ΔGdiss). Esto se debe a que el valor pKa es una medida de la constante de equilibrio para la reacción de disociación, y el cambio de energía libre de Gibbs es otra forma de expresar la constante de equilibrio.
El cambio de disociación de la energía libre de Gibbs (ΔGdiss) se puede predecir con métodos químicos cuánticos, como la teoría del funcional de la densidad (DFT) y la teoría de grupos acoplados (CC). Estos métodos proporcionan una descripción detallada de la estructura electrónica y las energías de las moléculas, lo cual es esencialpara predicciones precisas de ΔG.
Los métodos de química cuántica se pueden utilizar para calcular el cambio de energía libre de Gibbs de disociación simulando la reacción de disociación en un entorno mecánico cuántico. Esto implica calcular la energía de los reactivos, los productos y el estado de transición. A continuación, se calcula el cambio de energía libre de Gibbs a partir de la diferencia de energía entre los reactivos y los productos.
La teoría del funcional de la densidad (DFT) es un método ampliamente utilizado y computacionalmente eficiente para predecir propiedades moleculares. La DFT se ha utilizado con éxito para predecir la ΔGdiss de una variedad de moléculas. La precisión de las predicciones de ΔGdiss utilizando métodos químicos cuánticos puede mejorarse mediante el uso de teoría de alto nivel, como MP2 o CCSD(T). Sin embargo, estos métodos son más costosos desde el punto de vista computacional, lo que puede limitar su aplicación práctica a moléculas pequeñas. En general, la DFT es una buena opción para predecir la ΔGdiss para la mayoría de los ácidos y basesorgánicos.
Logros e hitos
Científicos del Departamento de Química Teórica de la Universidad del Ruhr de Bochum y BIOVIA determinaron los parámetros de LFER para su uso con la teoría COSMO-RS [1].
Se ha recogido un conjunto de 120 ácidos en el disolvente acetona para el que se dispone de datos experimentales de referencia. La predicción teórica de los valores de ΔGdiss se ha realizado con BIOVIA TURBOMOLE [2], COSMOconf [3] y COSMOtherm [4].
Para los ácidos orgánicos considerados en el trabajo, los ajustes de LFER arrojan muy buenas correlaciones lineales entre ΔGdiss calculado con COSMO-RS y DFT y los valores experimentales de pKa dentro de cada clase de compuesto.
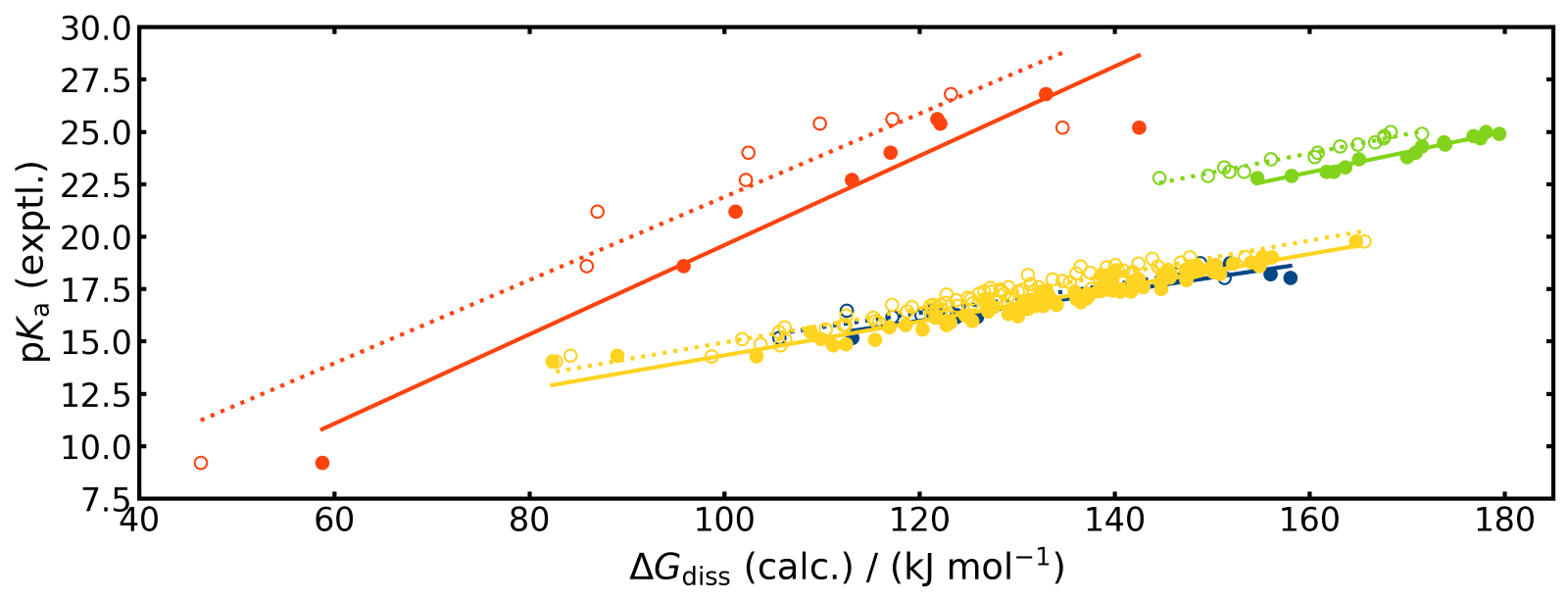
Figura 1: Se representa la relación lineal entre pKa y ΔGdiss.
Conclusión
La predicción precisa de los valores de pKa facilita la vida de los investigadores en muchos campos industriales. La aplicación de esta predicción se ha ampliado a los ácidos del disolvente acetona.
La predicción de los valores de pKa ácido en acetona utilizando COSMO-RS es una herramienta importante para comprender la reactividad química, diseñar productos farmacéuticos, desarrollar nuevos materiales, realizar estudios bioquímicos y realizar análisis computacionales eficientes. El flujo de trabajo establecido en esta investigación es muy prometedor como reemplazo de las mediciones experimentales no triviales y relativamente costosas de constantes de disociación ácida extremadamente bajas en solventes orgánicos
Referencias
- N. Sülzner, J. Haberhauer, C. Hättig, A. Hellweg, J. Comput. Chem. 2022, 43(15), 1011; https://doi.org/10.1002/jcc.26864
- TURBOMOLE V7.3, Un desarrollo de la Universidad de Karlsruhe y Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH 2007, 2018; http://www.turbomole.com.
- BIOVIA COSMOconf, Versión 2020; Dassault Systèmes. https://www.3ds.com/fileadmin/PRODUCTS-SERVICES/BIOVIA/PDF/BIOVIA-cosmoconf-datasheet.pdf .
- BIOVIA COSMOtherm, versión 2020; Dassault Systèmes. https://www.3ds.com/products/biovia/cosmo-rs/cosmotherm