
TURBOMOLE 2026
Programa de química computacional para simulaciones químicas cuánticas de moléculas, clústeres, sistemas periódicos y soluciones.
Descripción
Para los investigadores de todo el mundo, los cálculos de química cuántica son indispensables. Con TURBOMOLE®, un desarrollo de la Universidad de Karlsruhe y Forschungszentrum Karlsruhe GmbH (1989-2007) y TURBOMOLE GmbH (desde 2007), ofrecemos un paquete de programa de química cuántica muy potente y de uso general para cálculos de estructuras electrónicas ab initio con una amplia gama de aplicaciones.
BIOVIA TURBOMOLE incluye todos los métodos estándar y de última generación, un código DFT muy rápido para moléculas y sólidos, estados excitados y espectros que utilizan métodos DFT o de clúster acoplado. Los cálculos de alta precisión, así como algoritmos rápidos y de baja escala, y la paralelización permiten abarcar sistemas que antes estaban fuera de su alcance.
BIOVIA TURBOMOLE proporciona herramientas ultra eficientes y estables para simulaciones de química cuántica de moléculas, clústeres, sistemas periódicos y soluciones. Especializado en métodos con una relación precisión-coste excepcional, como la teoría funcional de densidad (DFT), incluida la aproximación de fase aleatoria (RPA), los métodos GW-Bethe-Salpeter, la teoría Møller-Plesset y los métodos de clúster acoplado explícitamente correlacionados.
Una de las principales aplicaciones de la química cuántica es la predicción exacta y la optimización de las reacciones químicas. Tareas como la optimización de catalizadores en entornos líquidos son un reto para los científicos de todos los sectores. Se requiere una combinación de diferentes métodos y pasos computacionales y TURBOMOLE ofrece una amplia variedad de herramientas y métodos para lograr la mejor precisión de su clase: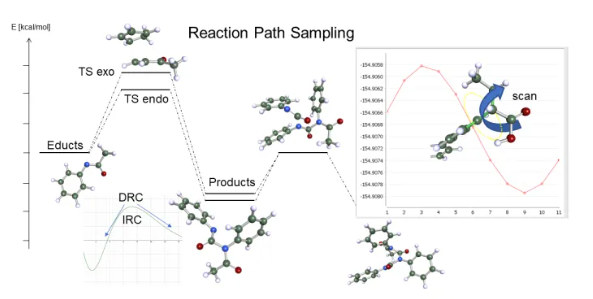
La espectroscopia es indispensable para el análisis de estructuras y el control de calidad en todos los campos de aplicación. Con BIOVIA TURBOMOLE, se pueden realizar predicciones y análisis de varios métodos, tanto para espectroscopia vibratoria como óptica: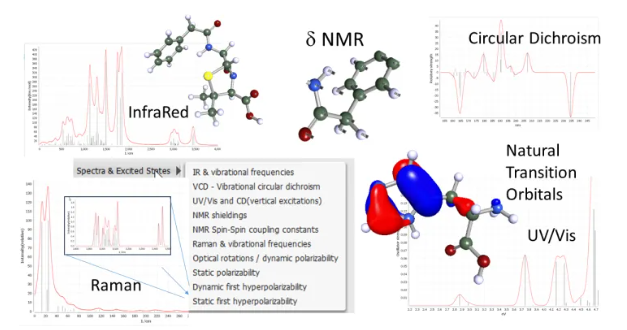
Los semiconductores orgánicos moleculares se pueden simular utilizando varios métodos basados en química cuántica para predecir las propiedades de las moléculas que emiten o absorben luz. BIOVIA TURBOMOLE ofrece en un flujo de trabajo automatizado propiedades como energías de reorganización, potenciales de oxidación/reducción o energía libre de Gibbs de solvación.
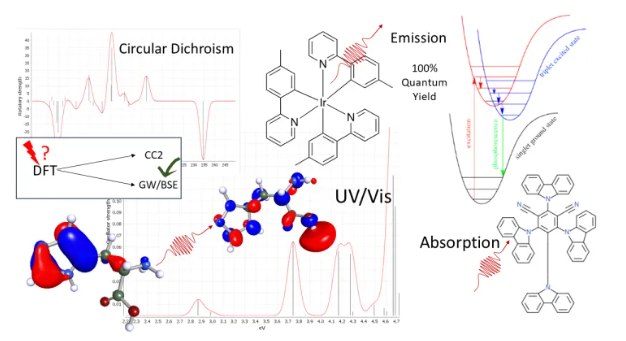
Los métodos de acoplamiento espín-órbita han sido pioneros en TURBOMOLE para elementos ligeros que utilizan cálculos relativistas exactos de todos los electrones, funciones DFT de última generación y GW de alta precisión, así como métodos de clúster acoplado que no sufren fallos típicos de DFT como excitaciones de transferencia de carga.
TmoleX es nuestra interfaz gráfica de usuario fácil de usar para gestionar rápidamente los cálculos de TURBOMOLE. Aunque tradicionalmente se han desarrollado conjuntos de química cuántica para un uso basado en línea de comandos o secuencias de comandos (para usuarios avanzados), TmoleX le permite utilizar química cuántica después de unos minutos de introducción. Es la herramienta perfecta para uso ocasional de TURBOMOLE que permite: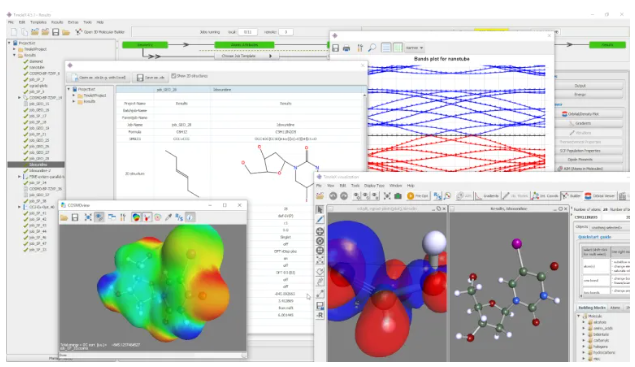
La nueva versión TURBOMOLE 2026 ya está disponible y está diseñada para científicos e investigadores en química computacional, química de solvatación y predicción espectroscópica. Esta actualización combina mejoras en parámetros de cálculo, interfaz de usuario y rendimiento, ofreciendo resultados más precisos y workflows más eficientes.
Descubre cómo TURBOMOLE 2026 puede acelerar tus proyectos de investigación y mejorar la calidad de tus simulaciones.
La versión más reciente de TURBOMOLE es la 7.9, que incluye numerosas nuevas funcionalidades y mejoras.
DESCRIPCIÓN
Modelado y simulación de moléculas y cristales
Para los investigadores de todo el mundo, los cálculos de química cuántica son indispensables. Con TURBOMOLE®, un desarrollo de la Universidad de Karlsruhe y Forschungszentrum Karlsruhe GmbH (1989-2007) y TURBOMOLE GmbH (desde 2007), ofrecemos un paquete de programa de química cuántica muy potente y de uso general para cálculos de estructuras electrónicas ab initio con una amplia gama de aplicaciones.
BIOVIA TURBOMOLE incluye todos los métodos estándar y de última generación, un código DFT muy rápido para moléculas y sólidos, estados excitados y espectros que utilizan métodos DFT o de clúster acoplado. Los cálculos de alta precisión, así como algoritmos rápidos y de baja escala, y la paralelización permiten abarcar sistemas que antes estaban fuera de su alcance.
Principales ventajas
- Resultados hasta un 80 % más rápidos
- Mejora del 25 % en la productividad de los investigadores
- Métodos de química cuántica computacional basados en la física
- Herramienta tanto de vanguardia como de última tecnología
- Potentes herramientas con las que pueden trabajar expertos y no expertos
BIOVIA TURBOMOLE proporciona herramientas ultra eficientes y estables para simulaciones de química cuántica de moléculas, clústeres, sistemas periódicos y soluciones. Especializado en métodos con una relación precisión-coste excepcional, como la teoría funcional de densidad (DFT), incluida la aproximación de fase aleatoria (RPA), los métodos GW-Bethe-Salpeter, la teoría Møller-Plesset y los métodos de clúster acoplado explícitamente correlacionados.
CARACTERÍSTICAS
REACCIONES
Una de las principales aplicaciones de la química cuántica es la predicción exacta y la optimización de las reacciones químicas. Tareas como la optimización de catalizadores en entornos líquidos son un reto para los científicos de todos los sectores. Se requiere una combinación de diferentes métodos y pasos computacionales y TURBOMOLE ofrece una amplia variedad de herramientas y métodos para lograr la mejor precisión de su clase:
- Optimizaciones de la ruta de reacción, búsquedas de estado de transición, escaneo de superficies de energía potencial
- Energías altamente precisas pero rápidas que utilizan estándares como CCSD(T) o RPA
- Efectos de solvación con una precisión inigualable gracias a COSMO-RS
- Altamente optimizado para hardware estándar, como estaciones de trabajo o clústeres multinúcleo
ESPECTROSCOPÍA
La espectroscopia es indispensable para el análisis de estructuras y el control de calidad en todos los campos de aplicación. Con BIOVIA TURBOMOLE, se pueden realizar predicciones y análisis de varios métodos, tanto para espectroscopia vibratoria como óptica:
- Espectros de infrarrojos (IR) y RAMAN
- Dicroísmo circular vibracional (VCD)
- UV-Vis óptico (absorción en espectros UV y visibles), CD (dicroísmo circular electrónico)
- Espectros de emisión y absorción vibrónica (SVL y VIPER)
DISPOSITIVOS ÓPTICOS
Los semiconductores orgánicos moleculares se pueden simular utilizando varios métodos basados en química cuántica para predecir las propiedades de las moléculas que emiten o absorben luz. BIOVIA TURBOMOLE ofrece en un flujo de trabajo automatizado propiedades como energías de reorganización, potenciales de oxidación/reducción o energía libre de Gibbs de solvación.
Los métodos de acoplamiento espín-órbita han sido pioneros en TURBOMOLE para elementos ligeros que utilizan cálculos relativistas exactos de todos los electrones, funciones DFT de última generación y GW de alta precisión, así como métodos de clúster acoplado que no sufren fallos típicos de DFT como excitaciones de transferencia de carga.
FÁCIL DE USAR
TmoleX es nuestra interfaz gráfica de usuario fácil de usar para gestionar rápidamente los cálculos de TURBOMOLE. Aunque tradicionalmente se han desarrollado conjuntos de química cuántica para un uso basado en línea de comandos o secuencias de comandos (para usuarios avanzados), TmoleX le permite utilizar química cuántica después de unos minutos de introducción. Es la herramienta perfecta para uso ocasional de TURBOMOLE que permite:
- Generación de entradas y visualizaciones de resultados
- Constructores moleculares 3D y 2D
- Varias funciones de importación y exportación
- Un gran número de métodos y propiedades
SECTORES
BIOVIA TURBOMOLE es utilizado por investigadores y profesionales en diversos sectores, incluyendo:- Industria química: Para el diseño y optimización de reacciones y catalizadores.
- Farmacéutica: En el desarrollo y análisis de compuestos bioactivos.
- Materiales: Para la investigación y desarrollo de nuevos materiales con propiedades específicas.
- Energía: En el estudio y mejora de dispositivos ópticos y semiconductores orgánicos.
VERSIONES
2026 (8.0)
La nueva versión TURBOMOLE 2026 ya está disponible y está diseñada para científicos e investigadores en química computacional, química de solvatación y predicción espectroscópica. Esta actualización combina mejoras en parámetros de cálculo, interfaz de usuario y rendimiento, ofreciendo resultados más precisos y workflows más eficientes.
Cálculos más robustos y confiables
TURBOMOLE 2026 incluye parámetros por defecto optimizados que permiten iniciar simulaciones con configuraciones más adecuadas, reduciendo errores y asegurando resultados consistentes en química cuántica.- Mayor estabilidad en cálculos DFT y COSMO
- Menos ajustes manuales requeridos
- Workflows más ágiles para investigadores
Predicción de espectros de alta calidad
La nueva versión mejora la predicción de espectros y otras propiedades moleculares, facilitando la comparación entre simulaciones teóricas y datos experimentales, un aspecto clave en investigación farmacéutica, química orgánica y materiales avanzados.Selección de solventes simplificada con COSMO
Gracias a la interfaz actualizada, TURBOMOLE 2026 permite seleccionar solventes de manera rápida y precisa para cálculos basados en COSMO, incluyendo efectos de solvatación y perfiles de carga en superficies. Esto acelera la preparación de simulaciones y reduce riesgos de errores.Mejoras internas para rendimiento y eficiencia
Las librerías backend y componentes internos se han actualizado para mejorar el rendimiento del software en cálculos de química cuántica compleja:- Optimización de motores de cálculo
- Mejor aprovechamiento del hardware
- Mayor estabilidad y rapidez en simulaciones largas
TURBOMOLE 2026: la herramienta completa para investigación en química computacional
Con estas mejoras, TURBOMOLE 2026 ofrece una combinación de precisión, eficiencia y usabilidad que facilita el trabajo diario de investigadores y científicos en química teórica, modelado molecular y simulación de materiales.Descubre cómo TURBOMOLE 2026 puede acelerar tus proyectos de investigación y mejorar la calidad de tus simulaciones.
7.9
La versión más reciente de TURBOMOLE es la 7.9, que incluye numerosas nuevas funcionalidades y mejoras.
Nuevas características
- ricc2:
- PTED-COSMO y PTED-PE para energías de excitación con CC2
- CCSDF12:
- Energías de excitación singlete CC3 (para una referencia Hartree-Fock de carcasa cerrada)
- Teoría de Incrustación Funcional de la Densidad (DFET)
- Partición del sistema en subsistemas activos y de entorno
- Tenga en cuenta los efectos ambientales a través del potencial de incorporación basado en DFT
- Incrustación de densidad congelada (FDE) o incrustación basada en proyección (PbE)
- Método de congelación y descongelación disponible
- DFT molecular incrustado en DFT periódica
- Incrustación periódica en periódica
- Métodos de función de onda (MP2, CCSD(T), ...) integrados en DFT
- Energías de excitación utilizando la Teoría de la Función de Onda (por ejemplo, CC2) incorporada en DFT
- Extensión a la Teoría
- Funcional de la Densidad Dependiente del Tiempo en Tiempo Real (RT-TDDFT) integrada en DFT
- Gradientes del conjunto de bases con respecto a los coeficientes de contracción en DFT
- Densidades de contacto Mössbauer y densidades de contacto efectivas con efectos relativistas y HF/DFT/RPA/MP2/CC2 arXiv 2407.21727
- Acoplamientos escalares-relativistas y no relativistas EPR hiperfinos en enfoques post-HF y post-KS DOI: 10.1021/acs.jpca.4c03794)
- Parámetros SNSO modificados del grupo Li (UW): enfoques universales y dependientes de la fila Dirac-Coulomb/Dirac-Coulomb-Breit DOI: 10.1021/acs.jctc.3c00479
- Enfoque DLU-X2C de baja escala, DLU modificado (NB), para todas las implementaciones relativistas (Energías, Gradientes, RMN, EPR, etc.)
- Se introdujo el modelo de carga gaussiana para COSMO para cavidades vdW con rejillas de Lebedev, lo que conduce a gradientes de geometría totalmente consistentes y hessianos DOI: 10.1021/acs.jctc.4c00052
- GOSTSHYP para simulaciones de alta presión, gradientes analíticos totalmente consistentes y estados excitados con HF/DFT
- Curvatura de Berry y cargas de Berry para cálculos en campos magnéticos finitos con HF/DFT
- Curvatura de Berry inducida por efectos relativistas (X2C) con HF
- Aproximación gaussiana gaussiana descongelada simple para simular espectros vibrónicos anarmónicos DOI:10.1021/acs.jctc.2c00030
- Respuesta no lineal de la ecuación de Bethe-Salpeter para la absorción de dos fotones y las hiperpolarizabilidades. (ESCF)
- Respuesta compleja ($damped_response) para respuesta no lineal dentro de TD-DFT y BSE. (ESCF)
- Cálculo de matrices T lineales (partes eléctricas y magnéticas) y no lineales (partes eléctricas) para excitaciones ópticas para simulaciones de materia ligera multiescala de vanguardia. (ESCF)
- Cálculo de matrices T lineales (partes eléctricas y magnéticas) y polarizabilidades para excitaciones vibratorias. (aoforce, NumForce)
- Nueva clase de aproximaciones funcionales de densidad altamente avanzadas basadas en interacciones generales de Fermiones: CHYF. El mejor rendimiento de su clase para TD-DFT, NMR y otras propiedades, junto con una excelente estabilidad numérica. (todos los módulos)
- DFT multicomponente (palabra clave $mcdft) que permite tratar otro fermión (por ejemplo, protones, muones, positrones ...) El tipo de Fermion es arbitrario, las correcciones relativistas están disponibles para cualquier Fermion. (RIDFT)
- Marco multicomponente de RPA y GW; es decir, se puede calcular la correlación y las energías de enlace de otro fermión a un sistema electrónico. (ESCF)
- Implementación inicial de DFT multicomponente dependiente del tiempo para calcular las interacciones luz-materia de otros fermiones en un sistema electrónico. (ESCF)
- Elementos de matriz de acoplamiento espín-órbita (SOCME) para TD-DFT y BSE entre estados fundamentales y excitados, así como entre estados excitados. (adecuado)
Mejoras
Eficiencia y facilidad de uso:- Mejora técnica en ricc2 y ccsdf12:
- Solucionador de valores propios no lineales rediseñado para ADC(2), CC2 y CC3
- Habilitar todas las opciones de usuario para X2C arXiv 2407.21727
- Se agregaron conjuntos de bases Dyall de elementos 3D y elementos de bloque s DOI: 10.5281/zenodo.7606547
- Asigne automáticamente jbas x2c-universal para todos los conjuntos de bases relativistas de electrones
- Soporte completo de la estimación inicial de la superposición de densidades atómicas en DOI más maduro: 10.1063/5.0209704
- Se agregaron nuevos funcionales híbridos locales LHJ-HFcal, TMHF, TMHF-3P, CHYF a TMoleX
- Se agregaron más parámetros de núcleo finito para la comparación con otros programas arXiv 2407.21727
- Se agregaron subenergías para funcionales híbridos locales a la salida
- Gráfico de la densidad de corriente en los cálculos de energía 2c DOI: 10.1063/5.0209704
- Agregue el término de contacto de Fermi estimado basado en la densidad en los núcleos solo en X2C
- Cribado aproximado mejorado para cálculos en campos magnéticos fuertes DOI: 10.1063/5.0217246
- Opciones para campos magnéticos externos en el programa de definición
- Aumento de LibXC a la versión 6.2.2.
- Experiencia de usuario mejorada para LibXC: Los funcionales ahora se pueden llamar usando su número O su nombre.
- Los derivados DFT 3rd se han reimplementado con grandes ganancias en eficiencia.
- Mejoras en el rendimiento general de senex. Use las pruebas de detección de manera más eficiente.
- Se ha mejorado el rendimiento de la GPU para GW y, especialmente, para BSE, incluida la compatibilidad con varias GPU para esta última.
- Se agregaron conjuntos de bases (orbitales y auxiliares) para protones cuánticos (def2-TZVPP-mc, def2-QZVPP-mc) para un excelente rendimiento dentro de los nuevos métodos DFT multicomponente.
- Se agregaron cuadrículas de integración adaptativas para DFT multicomponente.
- Nueva opción para cálculos RI-K TD-DFT completos en el núcleo, lo que acelera los casos en los que todos los intermedios caben en la memoria.
- $rigw variantes son más robustas cuando se tratan orbitales degenerados, lo que lleva a una menor ruptura de la simetría.
- Se agregaron opciones adicionales para la generación de TD-DFT NTO en propiedad.
- Se corrigieron los gradientes DFT-D4 para átomos ficticios
- La generación de orbitales de inicio mediante la superposición de densidades atómicas ahora funciona para todas las simetrías de grupos de puntos admitidas.
- Definir bloqueo en la sección RICC2 del menú general corregido
- impresión corregida de las energías totales de dispersión VV10 autoconsistentes
- Parámetros de dispersión corregidos para wB97X-D3
- Los trabajos ricc2 grandes que utilizan la paralelización MPI en varios nodos fallaron con el error de etiqueta MPI, corregido
- corregir el resultado de NaN para la corrección de carga periférica de COSMO en algunos casos especiales
7.8
Nuevas características
- DFT de dos componentes que utiliza condiciones de contorno periódicas (madurador de módulos) para efectos relativistas: energías, gradientes, tensor de tensión https://arxiv.org/abs/2305.03817
- Conjetura de SCF de dos componentes a partir de la superposición (no colineal) de densidades atómicas https://arxiv.org/abs/2305.03817
- TDDFT-ris: aceleración de dos órdenes de magnitud para cálculos híbridos TDDFT con un conjunto mínimo de bases auxiliares
- Funcionalidades ampliadas para la incrustación de densidad congelada (FDE):
- FDE rápido sin pasos supermoleculares en dscf y ridft, incluidas las versiones paralelas OMP y MPI
- FDE(ECP) para tener en cuenta la repulsión de Pauli
- Acoplamiento a RICC2 y PNOCCSD
- número arbitrario de https://doi.org/10.1063/5.0100393 de subsistema y https://doi.org/10.1021/acs.jctc.3c00347
- Teoría de la respuesta amortiguada CC2 (Complex Polarization Propagator) para el cálculo de espectros en regiones con alta densidad de estados
- función de respuesta lineal amortiguada para https://doi.org/10.1063/5.0042759 UV-vis y ECD
- función de respuesta cuadrática amortiguada para MCD y otros espectros inducidos https://doi.org/10.1021/acs.jctc.3c00536
- CC2 Faraday Término para los espectros de dicroísmo circular magnético (MCD) de moléculas con estados excitados degenerados, https://doi.org/10.1021/acs.jctc.3c00536
- Conversión automática de gradiente de campo eléctrico (EFG) a interacción de cuadrupolo nuclear (NQI) para EPR https://doi.org/10.1021/acs.jctc.1c01175
- Transformaciones de Euler EPR para tensor G, HFC, EFG, NQI
- Tensor g 1c con origen de gauge común para complementar los resultados de GIAO https://doi.org/10.1021/acs.jpca.2c03579
- Cambios de RMN de dos componentes con X2C/DLU-X2C, incluidos finnuc y mSNSO https://doi.org/10.1063/5.0171509
- Constantes de acoplamiento de RMN X2C/DLU-X2C escalares-relativistas, incluidas las https://doi.org/10.1021/acs.jctc.2c01248 finnuc
- Divisiones de campo cero, compatibles con el operador Pauli efectivo, SNSO o SOMF, X2C/DLU-X2C disponibles, incluido el https://doi.org/10.26434/chemrxiv-2023-2kh59-v2 finnuc
- Script hfacm: Compatibilidad mejorada con híbridos dobles y funcionales de modelos de conexión adiabática
- Se puede acceder al funcional híbrido local separado wLH22t (implementado para cálculos de DFT, TDDFT y gradiente de estado fundamental) mediante la palabra clave wlh22t.
- Tenga en cuenta que la versión 7.7 enlaza con parámetros diferentes a los que finalmente se publicaron https://doi.org/10.1021/acs.jctc.2c00782
- RIRPA: Gradiente de energía y analítico para varios funcionales Sigma https://doi.org/10.1063/5.0129524
- El nuevo híbrido local LH23pt con un rendimiento mejorado para las propiedades principales, implementado para energías, gradientes y TDDFT. https://doi.org/10.1002/jcc.27211
- Híbridos locales corregidos de fuerte correlación (scLH22ta, scLH22t, scLH23t-mBR, scLH23t-mBR-P) e híbridos locales separados por rangos corregidos de fuerte correlación (wLH23tE, wLH23tdE, wLH23tP, wLH23tdP, wLH23tB, wLH23tdB). https://doi.org/10.1063/5.0058917 , https://doi.org/10.1021/acs.jctc.2c00795 , https://doi.org/10.1063/5.0153463
- Métodos de ecuaciones de GW y Bethe-Salpeter que rompen la simetría de inversión en el tiempo, https://doi.org/10.1021/acs.jctc.3c00156
- Generación orbital virtual natural, principalmente para GW y BSE, https://doi.org/10.1063/5.0144469
- Evaluación de los estados ionizados y excitados del núcleo de TD-DFT y GW-BSE utilizando la aproximación CVS, https://doi.org/10.1063/5.0160265
- Métodos rápidos de deformación adaptativa del contorno de la rejilla GW (1c y 2c), https://doi.org/10.1063/5.0160265
- Parte mecánica cuántica de dispositivos fotónicos y modelado multiescala, https://doi.org/10.1002/adfm.202301093
- Respuesta cuadrática de la ecuación de Bethe-Salpeter
- Compatibilidad con cMGGA invariantes de calibre para gradientes de estado excitado y momentos dipolares en egrad y compatibilidad con MGGA y cMGGA invariantes de calibre para hiperpolarizaciones (dinámicas) y secciones transversales de absorción de dos fotones en https://doi.org/10.1021/acs.jctc.3c00259 de escf
Mejoras
- Rendimiento paralelo mejorado (OpenMP) de DFT periódica (madurador)
- Entrada COSMO simplificada (selección automática de la constante dieléctrica y el índice de refracción en función del nombre del disolvente para muchos disolventes)
- Agregue soscal en todas las características de órbita de espín en ridft, rdgrad, escf, mpshift, riper
- Desplazamiento orbital automático para SCF 2c (anteriormente se leía el automático pero se ignoraba, es decir, se usaba el desplazamiento de capa cerrada) https://arxiv.org/abs/2305.03817
- Superposición de densidades atómicas: ocupación de Hückel https://arxiv.org/abs/2305.03817
- Selección automática de isótopos para RMN y EPR según estándares experimentales
- Nuevo $epr de bandera para el cálculo simultáneo de todas las propiedades de EPR (HFC, G-TENSOR, EFG/NQI, ZFS) con opción de reinicio
- Compatibilidad con el paquete "treams" para la simulación de dispositivos multiescala y fotónicos https://github.com/tfp-photonics/treams/tree/main
- Algoritmos seminuméricos mejorados, cuadrículas pequeñas disponibles mejoradas
- Todas las funciones de GW y BSE ahora están totalmente disponibles en las GPU
- Conjuntos de bases:
- adición de conjuntos de bases pob-DZVP-rev2 y pob-TZVP-rev2 y las ECP correspondientes, útil para cálculos periódicos de DFT con madurador
- Conjuntos de bases Dyall para los elementos ligeros
- se agregaron todos los conjuntos básicos y ECP de Dolg
- Biblioteca de base reestructurada para PAE
- Fija:
- corregir la salida NICS para grandes distancias y corregir la redacción
- arreglar la salida de libxc con D3 en aoforce
- Se corrigió la entrada para métodos pseudoespectrales en MPSHIFT, redirige a Senex
- interfaz GIMIC mejorada, es decir, eliminar archivos que no son necesarios para GIMIC versión 2
- Se corrigieron las fugas de memoria para los gradientes LHF
- arreglar cuadrículas DFT con fullshell en mpshift
- corregir el uso simultáneo de $esenex y LHF en mpshift
- Corregir $intsdebug de palabras clave
- Se corrigieron las fugas de memoria en eVib
TmoleX
- Nuevo método GW y energías de excitación basadas en GW utilizando BSE (Ecuaciones de Bethe-Salpeter) disponibles en la interfaz gráfica de usuario
- La lista de archivos que se van a copiar de ejecuciones externas ahora se puede personalizar
- Al trabajar con varias pantallas que utilizan diferentes factores de escala, los usuarios experimentaron problemas con el tamaño incorrecto de los cuadros de diálogo y la falta de barras de desplazamiento. Estos problemas se han solucionado y la interfaz de usuario debería aparecer correctamente
- Las fuentes y los tamaños de fuente predeterminados de las interfaces de usuario de la aplicación Solvatation Chemistry se cambiaron para mejorar la legibilidad si se utiliza el escalado de la pantalla
- Las siguientes bibliotecas de terceros se han actualizado a una versión más reciente y/o con errores corregidos: 1. Biblioteca jogl de 2.4.0 a 2.5.0-rc 2. Biblioteca CDK de 2.7.1 a 2.8 4. Synthetica de 3.4.1 a 3.5.0 5. GSON de 2.9.0 a 2.10.1 6. MariadbClient de 2.7.2 a 3.1.4 7. Biblioteca SQLite 3.36.0.3 a 3.42.0.0 8. Biblioteca de puntos de interés de 5.2.2 a 5.2.3
7.5.1
Nuevas características
- Se han agregado las nuevas funciones: DFT r2-SCAN, r2-SCAN-3c y la dispersión DFT-D4 para r2-SCAN y r2-SCAN-3c. Información adicional.
- def-2-mTZVPP base establecida para r2-SCAN-3c. Información adicional.
- Se ha agregado también la función local DFT híbrida LH20-t a la lista de funciones conocidas. Información adicional.
- Conjuntos de base consistente de errores contratados segmentados de cuádruple ζ de valencia para cálculos relativistas de todos los electrones de uno y dos componentes.
Correcciones y mejoras
- Se ha agregado la corrección de carga periférica al usar la cavidad COSMO FINE para garantizar la estabilidad del algoritmo.
- Frecuencias vibratorias para cálculos DFT sin restricciones utilizando funciones MGGA con la biblioteca libxc habilitada nuevamente.
7.5
Nuevas características
DFT en general:
- Funcionales híbridos separados por rango en todos los módulos.
- Funcionales de correlación no local (VV10).
- Versión actualizada de LibXC (5.00 pre-versión).
Condiciones de contorno periódicas DFT:
- Energías de funciones híbridas (incluidas CAM-B3LYP y HSE separadas por rango).
Clúster acoplado (PNO, BCCD):
- Energías PNO-CCSD (T) de clúster acoplado.
- Energías de BCCD (T) -F12 de clúster acoplado de Brueckner correlacionadas para formas cerradas (RHF) y abiertas (UHF).
TDDFT, GW y Bethe-Salpeter:
- Respuesta amortiguada en TDDFT de uno y dos componentes y GW-BSE. Más información.
- Funcionales meta-GGA para gradientes TDDFT.
RPA:
- Energías totales del método de aproximación de fase aleatoria proyectada (RPA) semi-canónico generalizado de Kohn-Sham. Más información.
NMR (Apantallamientos y acoplamiento espín-espín):
- Constantes de acoplamiento espín-espín nuclear (todos los términos) en el ámbito de HF y DFT.
- Funcionales híbridos locales para apantallamiento químico de RMN. Más información.
Métodos y propiedades relativistas:
- Polarizabilidades de dos componentes (con corrección de cambio de imagen para DKH, BSS, X2C, incluido DLU). Más información.
- Cálculos de energía de dos componentes de un solo punto con campo eléctrico (con corrección de cambio de imagen para DKH, BSS y X2C, incluido DLU). Más información.
- Gradientes de conjuntos de bases X2C. Más información.
- Funcionales meta-GGA para cálculos de dos componentes.
Híbridos locales DFT:
- Híbridos locales de dos componentes para energías RI-DFT. Más información.
- Híbridos locales de dos componentes para estados con TDDFT excitados y espectros UV/Vis. Más información.
- Funcionales híbridos locales para gradientes TDDFT. Más información.
- Funcionales híbridos locales para apantallamientos químicos de RMN. Más información.
Espectro CD:
- Espectro ECD galga-invariante (dicroísmo electrónico circular).
Nuevos métodos:
- Campos magnéticos finitos dentro del método Hartree-Fock, con/sin RI-JK. Más información.
- Método Roothaan-Hall aumentado como solucionador de HF y DFT.
Nuevo módulo:
- Herramienta de predicción de color para espectros calculados y medidos.
Eficiencia
- Métodos pseudoespectrales para la energía y los gradientes TDDFT y para los cálculos de frecuencia vibratoria.
- Reducción de las demandas de memoria y el tiempo de cálculo en esquemas semi-numéricos para el cálculo del intercambio Hartree-Fock.
- Paralelización optimizada de OpenMP en todos los módulos, piezas XC reelaboradas y rutinas integrales (derivadas) de dos electrones.
- Convergencia mejorada para solucionador en mpshift.
- Enfoque de densidad diferencial en cálculos de dos componentes.
Usabilidad
- Primera versión de archivos de entrada simples (sin el uso de define) para flujos de trabajo automatizados o con secuencias de comandos.
- Fácil uso de apantallamientos químicos nucleares independientes NICS en todos los niveles de mpshift.
- Flujo de trabajo automatizado para el cálculo de funciones potenciales de reducción/oxidación.
- Nueva herramienta para predecir el color a partir de espectros calculados o medidos.
- TmoleX 4.6 ahora es compatible con:
- Constantes de acoplamiento Spin-Spin.
- Visualización de orbitales de transición usando TDDFT.
- Funcionales híbridos para energías de condición de contorno periódicas DFT.
- Cálculos de PNO-CCSD(T) sin restricciones.
- Advertencias de inconsistencias típicas durante la generación de entrada.