
BIOVIA Discovery Studio
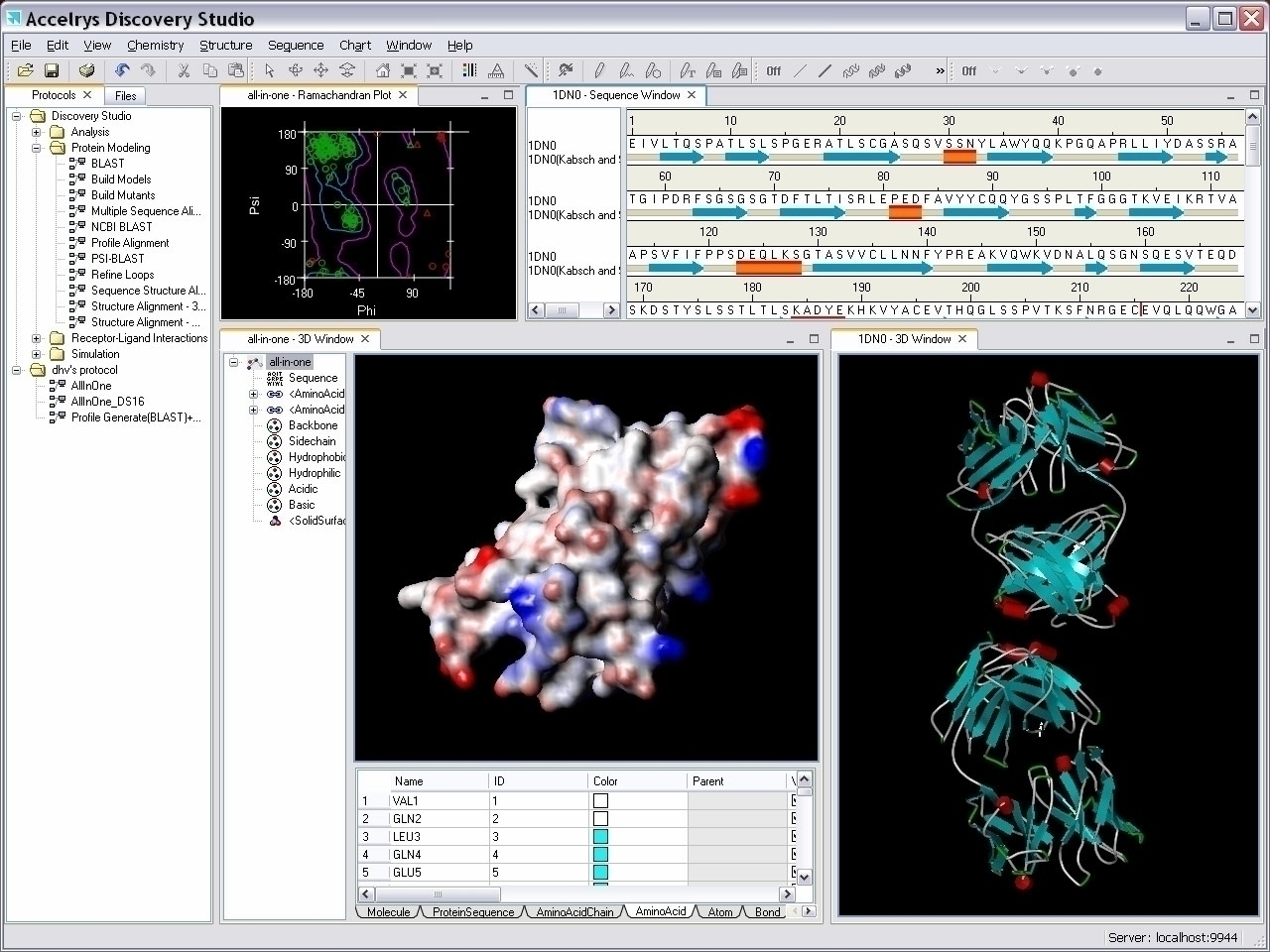
DESCRIPCIÓN
Modelado y simulación integrales para la investigación en ciencias de la vida
Las principales áreas en las que MS Modeling ofrece la tecnología mas avanzada en soluciones computacionales son:
- Modelado de Proteínas: hacer corresponder secuencias con patrones de estructura de proteínas, buscar en PDB, alinear secuencias, patrones o estructuras, y generar o analizar estructuras 3D. Estudiar las propiedades energéticas, dinámicas y electrostáticas en proteínas o en complejos de proteínas.
- Química Computacional/Química Médica: exploración de los modos de unión potenciales de nuevos ligandos mediante interacción y puntuación (docking and scoring), o extender la interacción con minimización in situ, puntuación y puntuación a librerías virtuales de compuestos buscando hits potenciales. Además, las herramientas de diseño de novo generan, encuentran y optimizan fragmentos de ligandos.
SECTORES
Simulaciones
Simule sistemas biológicos con las mejores herramientas de su clase

Los procesos biomoleculares se basan en una variedad de interacciones dinámicas entre proteínas, ligandos, solventes e iones. A menudo, los detalles de estas interacciones son difíciles de capturar solo a través de la experimentación física debido a las cortas escalas de tiempo en las que ocurren. La simulación puede ayudar a dilucidar la energía de estos procesos, proporcionando información sobre su mecanismo de acción y propiedades.
BIOVIA Discovery Studio utiliza los mejores programas de simulación molecular de su clase, NAMD y CHARMm. Además, la dinámica molecular acelerada gaussiana (GaMD) también se implementa en la última versión de Discovery Studio para el muestreo mejorado sin restricciones simultáneo y los cálculos de energía libre.
Simular
- ENCANTOm
- NAMD
- Realización de simulaciones explícitas de MD con disolventes
- Solvate una proteína con membrana explícita y ejecuta simulaciones de MD
- DMol3 / CHARMm
- Calcule energías de un solo punto o realice minimizaciones de complejos receptor-ligando utilizando simulaciones híbridas de Mecánica Cuántica/Mecánica Molecular (QM/MM)
- Implementación de GaMD para el muestreo mejorado sin restricciones simultáneo y los cálculos de energía libre
- Configure y ejecute un equilibrio de GaMD, parametrizando automáticamente los potenciales de refuerzo necesarios
- Ejecución y reinicio de simulaciones de GaMD
- Estime un panorama de energía libre a partir de un conjunto de trayectorias de MD, lo que permite la reponderación estadística de las simulaciones de GaMD
Modelo
- Soporte para una amplia gama de campos de fuerza, incluidos CGenFF, charmm36, CHARMm y más
- Método MATCH para tipificar ligandos con charmm36
- Soporte completo del mecanismo de aplicación de parches CHARMM
- Método de solvatación acuosa explícita rápida con contraiones opcionales adecuado para sistemas moleculares muy grandes
- Solvatación de proteína transmembrana en bicapa lipídica preequilibrada
- Análisis de las trayectorias de la DM
Explorar
- Realice predicciones rápidas y precisas de ionización de proteínas y pKs de residuos para la preparación de proteínas
- Utilice CDOCKER, un motor de acoplamiento basado en CHARMm, para realizar un acoplamiento y refinamiento flexibles basados en ligandos
- Realizar la optimización de la pose de múltiples ligandos en el contexto de un receptor
- Calcular las energías de enlace de las posturas acopladas
- Predecir con precisión la energía relativa de unión del ligando para una serie de ligandos congenéricos utilizando el método de perturbación de energía libre (FEP)
- Calcular la energía libre relativa de unión para una biblioteca combinatoria de ligandos modelada por Multi-Site Lambda Dynamics (MSLD)
- Estime la energía libre de unión al ligando y estudie la unión del ligando mediante simulaciones de dinámica molecular dirigida (SMD) basadas en CHARMm
- Examine los efectos del potencial electrostático con la ecuación CHARMm Poisson-Boltzmann (PB)
Diseño basado en la estructura
Facilite el descubrimiento de fármacos, desde el descubrimiento de aciertos hasta la optimización en las últimas etapas

El Diseño Basado en Estructuras (SBD, por sus siglas en inglés) y el Diseño Basado en Fragmentos (FBD, por sus siglas en inglés) son estrategias bien establecidas en el desarrollo racional de fármacos de moléculas pequeñas. El conocimiento de cómo una molécula pequeña se une a una proteína ofrece ventajas considerables, tanto en términos de priorizar compuestos para la detección en etapas tempranas, como de optimizar la potencia y la selectividad.
BIOVIA Discovery Studio ofrece una cartera completa y escalable de herramientas científicas, incluido GOLD del Centro de Datos Cristalográficos de Cambridge (CCDC), para respaldar y ayudar a las estrategias SBD y FBD desde el descubrimiento de aciertos hasta la optimización de clientes potenciales en etapa tardía.
Preparar
- Analizar y preparar estructuras 3D (p. ej., PDB, estructura de rayos X, modelo de homología) para SBD
- Construya automáticamente moléculas vecinas basadas en el empaquetamiento de cristales y analice sus interacciones
- Predicción de los estados de ionización de los residuos al pH elegido
- Identificar y estudiar los sitios putativos de unión de ligandos
- Prepare ligandos con un amplio conjunto de características y calcule coordenadas 3D
- Generar conformaciones de ligandos
- Filtrar ligandos en función de la similitud con los fármacos, las propiedades moleculares o para eliminar grupos o características no deseados
Pantalla
- Identificación y optimización de visitas
- Realice un cribado virtual de ligandos y fragmentos utilizando el motor de farmacoforo CATALYST o los enfoques de acoplamiento LibDock o CDOCKER
- Realizar el acoplamiento con GOLD §
- Realice la optimización de plomo in situ utilizando transformaciones de reacción de química médica clásica y reactivos disponibles comercialmente
- Salto de andamio o sustitución del grupo R in situ utilizando fragmentos moleculares derivados de compuestos disponibles comercialmente
§ Requiere licencia del Centro de Datos Cristalográficos de Cambridge
Puntuación
- Calcule las energías de enlace con métodos basados en MM-PBSA o MM-GBSA CHARMm
- Predecir con precisión la energía relativa de unión del ligando para una serie de ligandos congenéricos utilizando el método de perturbación de energía libre (FEP)
- Calcular la energía libre relativa de unión para una biblioteca combinatoria de ligandos modelada por Multi-Site Lambda Dynamics (MSLD)
- Identifique los residuos críticos que interactúan utilizando un conjunto completo de monitores favorables, desfavorables e insatisfechos que no son adherentes
- Perfilar y priorizar los resultados de las pruebas de detección, optimizando la potencia y la especificidad del objetivo
Extender
- Diseñe y optimice bibliotecas combinatorias como nuevos puntos de partida para su posterior cribado.
- Combine sus puntuaciones con QSAR clásico, huellas dactilares y descriptores basados en la mecánica cuántica y cree modelos predictivos avanzados
- Minimice la toxicidad con TOPKAT y optimice el perfil farmacocinético.
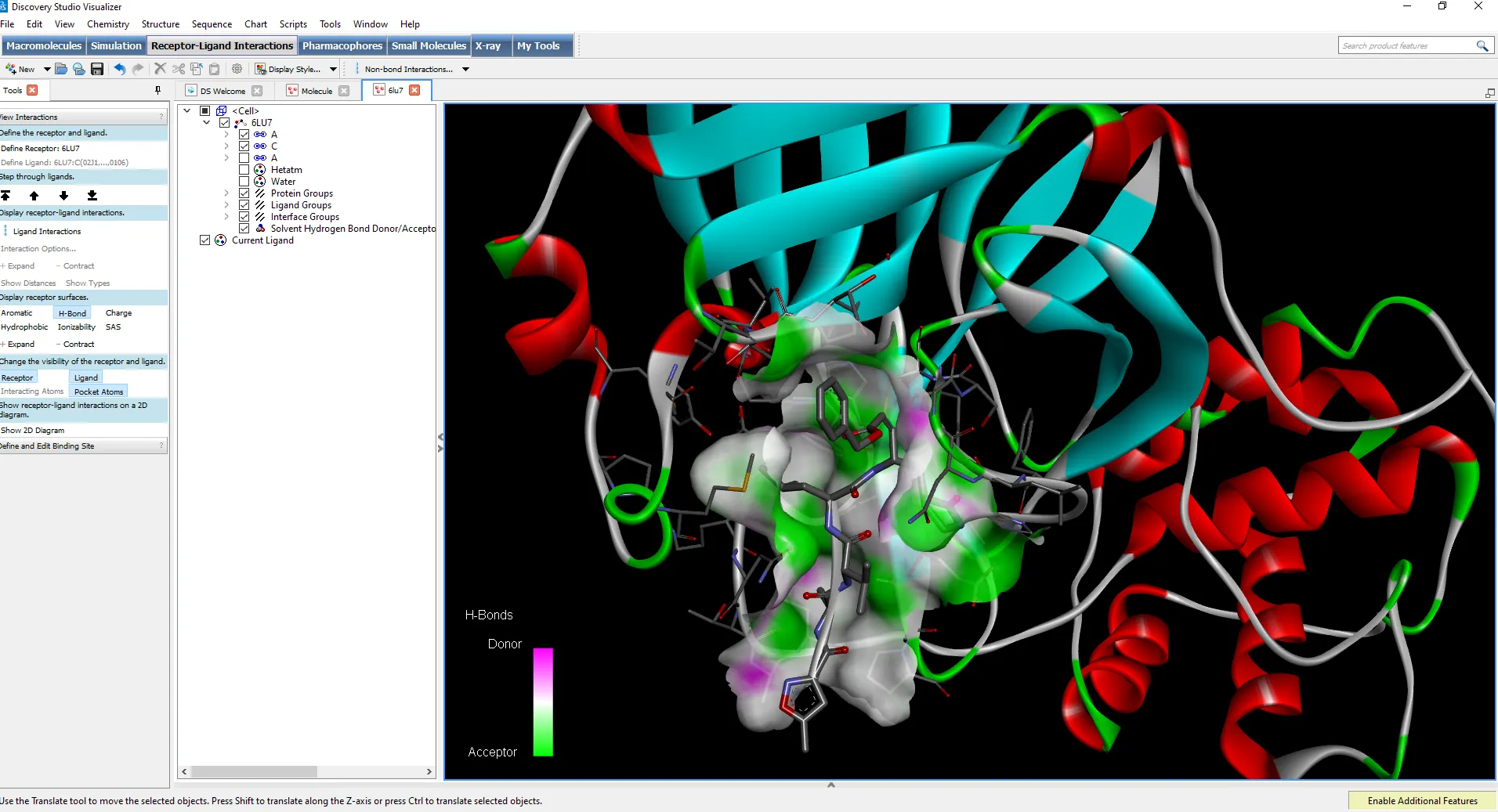
Diseño basado en ligandos y farmacóforos
Mejore el diseño terapéutico de moléculas pequeñas con las mejores herramientas de su clase
Las interacciones de unión de proteínas y ligandos surgen de una variedad de factores estéricos y electroquímicos. Comprender estas interacciones puede ayudar a los investigadores a identificar más rápidamente nuevos candidatos terapéuticos prometedores. Los farmacóforos mapean estas características en el espacio 3D, proporcionando un medio simple para examinar bibliotecas de compuestos virtualmente, caracterizar la unión de los cables existentes y optimizar su rendimiento.
BIOVIA Discovery Studio utiliza el conjunto de herramientas de modelado y análisis de farmacoforos CATALYST para ayudar en la evaluación de terapias de moléculas pequeñas con o sin datos estructurados en el objetivo. Es compatible con el diseño de fármacos de novo, el diseño de fármacos multiobjetivo y el perfil de actividad para impulsar la investigación y el desarrollo de moléculas pequeñas.
Construir
- Genere automáticamente farmacóforos a partir de los datos disponibles
- Conjuntos de ligandos activos
- Sitios de unión al receptor
- Complejos receptor-ligando
- Realizar una rigurosa validación de farmacóforos basada en conjuntos de compuestos de control con actividad conocida.
- Las hipótesis pueden incluir:
- Consultas geométricas basadas en características
- Similitud de forma
- Espacio "prohibido"
- Vaya más allá de las limitaciones de los algoritmos clásicos de elucidación de farmacóforos explorando los farmacóforos de conjunto para conjuntos de compuestos muy grandes/diversos con un riesgo de múltiples modos de acción
Aplicar
Llevar a cabo estudios sólidos de detección de farmacoforos
- Construir y buscar bases de datos de conformaciones 3D
- Considere y analice el espacio conformacional completo de sus ligandos
- Explore la actividad fuera del objetivo y la reutilización de fármacos utilizando la base de datos PharmaDB*
Discovery Studio ahora incluye la base de datos más extensa reportada para la creación de perfiles de ligandos. Construido y validado a partir del scPDB*, el PharmaDB contiene aproximadamente 240.000 modelos de farmacoforos receptores-ligandos.
Diseño
Diseñe y caracterice sus ligandos y bibliotecas combinatorias
- Enumeración de bibliotecas basadas en reacciones o núcleos
- Enumerar estados de ionización, tautómeros e isómeros
- Filtre a los candidatos pobres con grupos funcionales indeseables y reglas de Lipinski y Veber o sus propios criterios
- Calcule numerosas propiedades fisicoquímicas y de huellas dactilares
- Optimice las bibliotecas combinatorias mediante la optimización de Pareto, el análisis de diversidad y similitud
- Herramientas de clustering y visualización 3D mediante análisis PCA
Bioterapéutica y modelado de anticuerpos
Mejorar el diseño y el desarrollo de productos bioterapéuticos
Los bioterapéuticos ofrecen una gama de beneficios únicos sobre los medicamentos de moléculas pequeñas. Su mayor afinidad y selectividad han permitido a los investigadores abordar objetivos nuevos y difíciles. Esto ha llevado a un aumento dramático en los proyectos de investigación y desarrollo centrados en anticuerpos y otras modalidades biológicas para el tratamiento, como los biespecíficos. Sin embargo, estos proyectos también deben superar retos similares en materia de seguridad y farmacocinética a los que se enfrentan las moléculas pequeñas, aunque desde una perspectiva diferente. Factores como la alta inmunogenicidad o la baja solubilidad pueden acabar con el desarrollo de un candidato que, por lo demás, es eficaz.
El modelado y la simulación pueden avanzar significativamente en el desarrollo de estas nuevas clases de terapias. Por ejemplo, los investigadores pueden predecir las propiedades de formulación de un anticuerpo y sugerir mutaciones para mejorarlas rápidamente y a bajo costo en comparación con la experimentación de laboratorio solamente. BIOVIA Discovery Studio ofrece un amplio conjunto de herramientas para ayudar a guiar el diseño de bioterapias, lo que permite a los equipos optimizar el rendimiento de los candidatos in silico y agilizar su trabajo físico.
Modelo
- Cascada de modelado automatizado para generar fácil y rápidamente modelos 3D de anticuerpos de longitud completa, Fab o Fv de alta calidad a partir de un conjunto de secuencias de cadenas de anticuerpos ligeros y pesados y una base de datos de plantillas de anticuerpos PDB seleccionada
- Compatibilidad total con los esquemas de anotación de anticuerpos de uso común: IMGT, Chothia, Kabat y Honegger
- Realice de forma simultánea e independiente múltiples alineaciones de secuencias de cadenas pesadas y ligeras
- Herramientas expertas adicionales para identificar estructuras de plantillas de anticuerpos y construir modelos de homología de alta calidad
- Capacidad para construir estructuras biespecíficas de anticuerpos de longitud completa
- Perfeccione los bucles CDR mediante plantilla, injerto de bucle o modelado de bucle de novo
- Realizar análisis detallados del modelo
Diseño
- Realizar predicciones de afinidad de unión y estabilidad mutacional térmicas o basadas en el pH
- Identificar posibles ubicaciones estables de puentes disulfuro
- Realice el acoplamiento anticuerpo-antígeno utilizando ZDOCK para identificar residuos críticos que interactúan
- Predecir las mutaciones de los residuos para la humanización de los anticuerpos sin comprometer la estabilidad o la eficacia de los anticuerpos
- Estudie la flexibilidad conformacional con simulaciones explícitas de dinámica molecular (MD) basadas en disolventes utilizando CHARMm o NAMD
Predecir
- Calcule las propiedades biofísicas, incluido el punto isoeléctrico, la solubilidad, la viscosidad (SCM*) y la propensión a la agregación (índice de desarrollabilidad – SAP*) para la evaluación de la idoneidad para el desarrollo en las primeras etapas.
- Prediga sitios propensos a modificaciones postraduccionales (PTM) mediante la búsqueda de motivos basada en secuencias
*Con licencia del MIT
Diseño y análisis de macromoléculas
Mejorar la investigación basada en macromoléculas
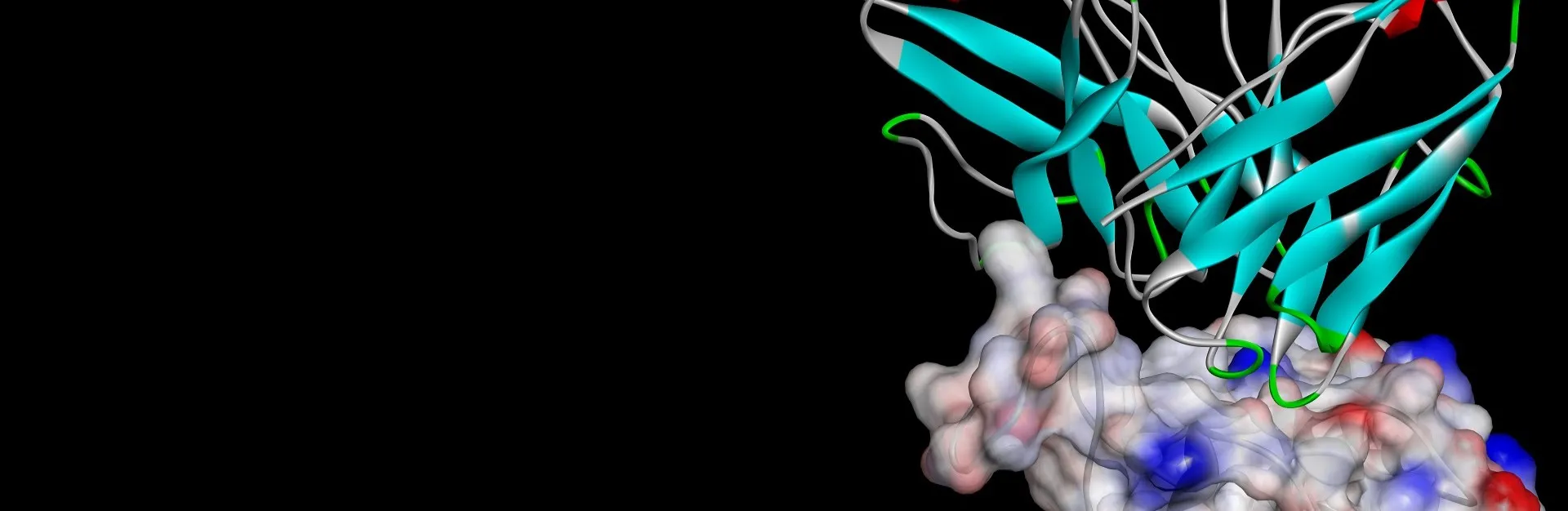 La determinación de la estructura tridimensional y las propiedades de las macromoléculas, como las enzimas y los anticuerpos, es un componente fundamental para una amplia gama de actividades de investigación. Por ejemplo, las diferentes conformaciones que surgen de la dinámica molecular normal o de las interacciones con ligandos u otras proteínas pueden revelar nuevos sitios de unión o proporcionar pistas sobre su función. Si bien miles de moléculas han resuelto experimentalmente sus estructuras, la obtención de datos estructurales de alta fidelidad sigue siendo un proceso no trivial.La simulación puede aumentar la experimentación física al proporcionar información sobre la estructura macromolecular. Además, técnicas como el modelado de homología pueden ayudar a predecir modelos estructurales para moléculas novedosas, guiando el diseño terapéutico y los esfuerzos de ingeniería de proteínas. BIOVIA Discovery Studio ofrece una cartera completa de herramientas científicas validadas líderes en el mercado, capaces de ayudar en todos los aspectos de la investigación basada en macromoléculas.
La determinación de la estructura tridimensional y las propiedades de las macromoléculas, como las enzimas y los anticuerpos, es un componente fundamental para una amplia gama de actividades de investigación. Por ejemplo, las diferentes conformaciones que surgen de la dinámica molecular normal o de las interacciones con ligandos u otras proteínas pueden revelar nuevos sitios de unión o proporcionar pistas sobre su función. Si bien miles de moléculas han resuelto experimentalmente sus estructuras, la obtención de datos estructurales de alta fidelidad sigue siendo un proceso no trivial.La simulación puede aumentar la experimentación física al proporcionar información sobre la estructura macromolecular. Además, técnicas como el modelado de homología pueden ayudar a predecir modelos estructurales para moléculas novedosas, guiando el diseño terapéutico y los esfuerzos de ingeniería de proteínas. BIOVIA Discovery Studio ofrece una cartera completa de herramientas científicas validadas líderes en el mercado, capaces de ayudar en todos los aspectos de la investigación basada en macromoléculas.Buscar
- Realice múltiples búsquedas de secuencias utilizando BLAST y PSI-BLAST en bases de datos locales o NCBI
- Para proteínas de cadena múltiple, realice de forma simultánea e independiente múltiples alineaciones de secuencia de cada cadena de proteína
- Predicción de las hélices transmembrana en secuencias de proteínas transmembrana
- Prediga sitios propensos a modificaciones postraduccionales (PTM) mediante la búsqueda de motivos basada en secuencias
Modelo
- Analizar y preparar estructuras a partir de repositorios de estructuras 3D (por ejemplo, PDB)
- Generación de modelos de estructura 3D con MODELER
- Verificar la calidad de un modelo de estructura
- Utilice LOOPER para buscar sistemáticamente conformaciones de bucle y clasificar usando CHARMm
- Injertar conformaciones de bucle de una estructura de plantilla en un modelo de destino
- Optimice sistemáticamente las cadenas laterales de aminoácidos utilizando las simulaciones de ChiRotor CHARMm
- Utilice ZDOCK para realizar el acoplamiento proteína-proteína y examinar las interacciones de los socios de unión
- Estudie la flexibilidad conformacional con simulaciones explícitas de dinámica molecular (MD) basadas en disolventes utilizando CHARMm o NAMD
Diseño
- Predecir las propiedades eléctricas de la proteína, incluida la estabilidad dependiente del pH y los estados de protonación y el punto isoeléctrico
- Realizar predicciones de afinidad de unión y estabilidad mutacional térmicas o basadas en el pH
- Identificar posibles ubicaciones estables de puentes disulfuro
- Calcular las propiedades biofísicas importantes para la formulación de proteínas, incluidas la viscosidad y la solubilidad
QSAR, ADMET y Toxicología Predictiva
Descubra terapias seguras y eficaces
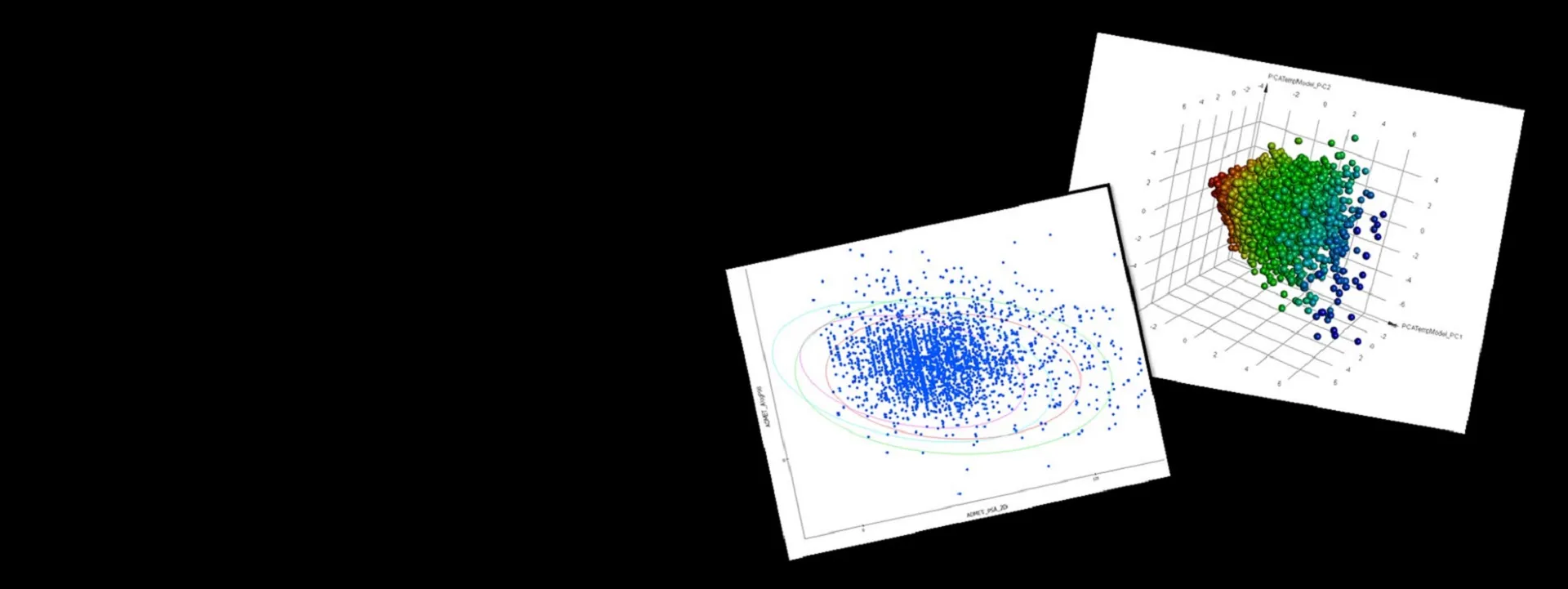
Comprender y cuantificar las relaciones estructura-actividad puede tener un impacto significativo en la optimización de clientes potenciales y el desarrollo de fármacos al minimizar la experimentación tediosa y costosa. Construya, valide y aplique sus propios modelos basados en una amplia gama de enfoques, y siga mejorándolos a medida que se disponga de nuevos datos. Evalúe el riesgo potencial que plantean las propiedades farmacocinéticas desfavorables y la toxicidad potencial utilizando los modelos distribuidos de BIOVIA Discovery Studio, amplíelos para cubrir mejor su espacio químico patentado y utilice las indicaciones comprensibles para sortear dichos pasivos.
QSAR
- Preparación de datos completa y coherente:
- Ligandos: eliminan duplicados y manejan tautómeros e ionización
- Preparar la propiedad de respuesta (escalado y discretización)
- Divida los datos en conjuntos de entrenamiento y prueba con los métodos adecuados
- Elija entre una gran cantidad de fisicoquímicos, topológicos, electrónicos, geométricos. descriptores basados en la huella dactilar y la mecánica cuántica
- Cree modelos estadísticos que incluyan Bayesian, MLR (Regresión Lineal Múltiple), PLS (Mínimos Cuadrados Parciales) y GFA (Análisis Funcional Genético)
- Analice y valide modelos mediante dominios de aplicabilidad de modelos (MAD), validación automática de conjuntos de pruebas, validación cruzada y métricas estadísticas
- Identificar las transformaciones de los pares moleculares emparejados (MMP) y estudiar los acantilados de actividad
ADMET
Obtenga una evaluación temprana de sus compuestos calculando las propiedades ADMET (absorción, distribución, metabolismo, excreción y toxicidad) previstas para colecciones de moléculas como candidatos a síntesis, bibliotecas de proveedores y colecciones de cribado. Utilice los resultados para eliminar compuestos con características ADMET desfavorables y evalúe los refinamientos estructurales propuestos, diseñados para mejorar estas propiedades antes de la síntesis.Los descriptores de ADMET incluyen:
- Absorción intestinal humana
- Solubilidad acuosa
- Penetración de la barrera hematoencefálica
- Unión a proteínas plasmáticas
- Unión al CYP2D6
- Hepatotoxicidad
- Filtrar conjuntos de moléculas pequeñas para grupos de funciones no deseados basados en las reglas publicadas de SMARTS
Toxicidad
Evalúe el rendimiento de sus compuestos en ensayos experimentales y modelos animales. Calcule y valide las evaluaciones de los efectos tóxicos y ambientales de los productos químicos únicamente a partir de su estructura molecular. TOPKAT® (TOxicity Prediction by Komputer Assisted Technology) emplea modelos robustos y validados de relación de toxicidad de estructura cuantitativa (QSTR) para evaluar varios puntos finales y utiliza el método patentado de validación de espacio predictivo óptimo para ayudar a interpretar los resultados.Nota:
Los detalles sobre nuestros modelos extensibles TopKat se han publicado en formato de informe de modelo QSAR (QMRF) en la "Base de datos de modelos QSAR" del Centro Común de Investigación (JRC) de la Comisión Europea.
- Mutagenicidad de Ames
- Carcinogenicidad de roedores (datos de NTP y FDA)
- Peso de la evidencia carcinogenicidad
- Potencia cancerígena TD50
- Potencial de toxicidad para el desarrollo
- Rata oral DL50
- Dosis máxima tolerada en ratas
- Toxicidad por inhalación de ratas LC50
- Rata crónica LOAEL
- Irritación y sensibilización de la piel
- Irritación ocular
- Biodegradabilidad aeróbica
- Pececillo cabeza gorda LC50
- Daphnia magna EC50
Visualización
Visualiza tu ciencia
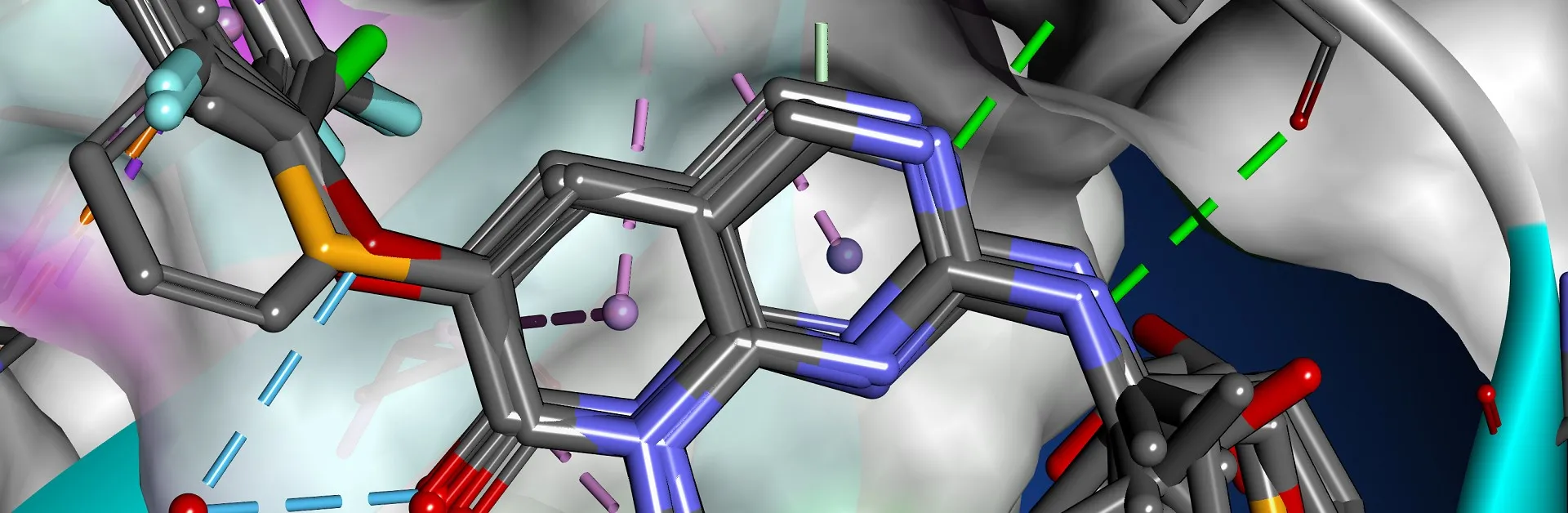
BIOVIA Discovery Studio Visualizer es una aplicación de modelado molecular gratuita y rica en funciones para ver, compartir y analizar datos de proteínas y moléculas pequeñas. Los expertos y sus colegas pueden intercambiar resultados de manera fluida y eficiente, sin pérdida de tiempo ni de información científica.
El visualizador de Discovery Studio proporciona una colección completa de funciones para capturar los matices específicos de su investigación:
Capacidades generales
- Gráficos de alta calidad con opciones de visualización avanzadas y soporte estéreo
- Posibilidad de generar imágenes de calidad de publicación
- Vista gráfica interactiva en 3D con jerarquía asociada y vistas de tabla de datos
- Múltiples superficies e iso-superficies laterales para mejorar la visualización molecular
- Capacidad para trazar datos de múltiples series de datos con gráficos de líneas y puntos, gráficos 3D y gráficos de barras
- Capacidad de gráficos que incluye mapas de calor, histogramas, gráficos de tasa de aciertos y más
- Funcionalidad de guion gráfico para capturar series de vistas moleculares para demostrar y compartir
- Exportar guiones gráficos como películas
- Funcionalidad de ordenación, filtrado y agrupación disponible para las propiedades de la tabla de datos
- Secuencias de comandos de Perl para automatizar tareas repetitivas o vincular tareas para facilitar su uso
- Los paneles de herramientas, las barras de herramientas y el diseño de la interfaz se pueden personalizar como se desee
- Compatibilidad con una amplia variedad de formatos de archivo de estructura y secuencia
- Cálculos RMS disponibles en diferentes niveles de detalle
- Monitores disponibles para una amplia variedad de interacciones favorables, desfavorables e insatisfechas sin vínculo
Diseño y visualización de proteínas
- Constructores moleculares para péptidos y ácidos nucleicos
- Cálculo de la accesibilidad a los disolventes para identificar los residuos enterrados y expuestos
- Funcionalidad de gráficos, incluidos Ramachandran y gráficos de contacto
- Cargue estructuras de proteínas directamente desde la base de datos de PDB
- Genere informes de proteínas para resumir los datos en las estructuras de proteínas
- Construir la unidad biológicamente activa de una proteína a partir de sus subunidades
- Limpie la funcionalidad de las proteínas para agregar cadenas laterales faltantes, eliminar el desorden y estandarizar la nomenclatura de los átomos
- Visualización mejorada de secuencias de proteínas y ácidos nucleicos y análisis de su composición y alineaciones
- Superposición de estructuras basadas en ataduras, residuos y alineación de secuencias
- Predicción de la estructura secundaria de secuencias de proteínas
- Ventana de anotaciones para ver y editar anotaciones de secuencias de ácidos nucleicos y proteínas
- Visualización y contorno de mapas de densidad de electrones de rayos X
- Herramientas básicas para editar estructuras de rayos X
- Muestreo de conformaciones de rotámeros de cadena lateral y análisis de interacción
Diseño y visualización de ligandos
- Superficies de interacción receptor-ligando que muestran hidrofobicidad, enlaces de hidrógeno, aromaticidad y más
- Ver representación 2D de moléculas en la tabla de datos
- Herramientas de boceto para crear nuevas moléculas pequeñas
- Modifique o construya moléculas pequeñas en 3D personalizadas utilizando un panel de herramientas de fragmentos predefinidos
- Optimice la geometría de las estructuras construidas con un campo de fuerza rápido similar al de Dreiding
- Se pueden calcular propiedades moleculares básicas, como la fórmula molecular y el peso molecular
- Superposición de estructuras basadas en la superposición molecular mediante alineación de campo o anclajes
- Defina, muestre y edite sitios de unión de ligandos
- Generar gráficos de interacción receptor-ligando en 2D
- Analizar los patrones de unión de ligandos entre una proteína y sus ligandos unidos
- Generación manual de consultas de farmacóforos (catalizador)
CARACTERÍSTICAS
Plataforma
Discovery Studio Standalone
Completa plataforma de modelado molecular diseñada para el modelador independiente. Este entorno “standalone”, impulsado por la plataforma abierta Pipeline Pilot, incluye toda la infraestructura necesaria para diseñar y llevar a cabo experimentos con Discovery Studio Science: visualizar, modelar y analizar datos biológicos y químicos de forma sencilla, empleando herramientas para dibujar moléculas 3D, visualizar cambios dinámicos, realizar representaciones 3D y alojar otras muchas funcionalidades. La instalación “standalone” puede también conectarse a Pipeline Pilot Server para compartir fácilmente datos y flujos de trabajo.Discovery Studio Visualizer Client
Potente interfaz gráfica para acceder a Discovery Studio Science. DS Visualizer Client se puede instalar y mantener de forma sencilla para grupos de investigación sin renunciar a prestaciones de primera clase. Si se implementa con Pipeline Pilot Server, permite capacidades inigualables para compartir datos, flujos de trabajo y recursos computacionales. Pipeline Pilot Server
Obtiene el máximo rendimiento de su información a través del control de flujo de datos de escala industrial y de sus potentes capacidades de extracción. Permite componer gráficamente redes de procesamiento de datos, con cientos de diferentes componentes configurables para operaciones como recuperación, manipulación, filtrado y visualización. Estos protocolos se capturan automáticamente a medida que los crea, y puede incluso publicarlos para su uso a nivel corporativo. Sus colaboradores tienen la opción de solicitar sus protocolos y ejecutarlos con sus datos propios usando una simple interfaz web. Discovery Studio Free Visualizer
Muestra y comparte datos de proteínas y de moléculas pequeñas de forma clara y consistente, ofreciendo una amplia variedad de formatos estándar. Esta herramienta de visualización gratuita y fácil manejo es una solución ideal para investigadores que colaboran con modeladores, pero no necesitan acceder a las herramientas de análisis a nivel de expertos de Discovery Studio.
Herramientas para modelado de proteínas
| HERRAMIENTA | DESCRIPCIÓN |
| DS Modeler | Genera rápidamente, de forma automática, un modelo de homología refinado a partir del alineamiento de secuencia con una proteína de estructura 3D conocida |
| DS Protein Refine | Optimiza regiones loop de una proteína usando un algoritmo propio basado en CHARMm |
| DS Protein Families | Analiza los patrones de conservación de secuencia y la posición de los residuos conservados en la estructura 3D dentro de las secuencias de una familia de proteínas |
| DS Protein Health | Revisa la validez y calidad de la estructura de una proteína (o parte de la estructura) obtenida a partir de modelado o de datos experimentales |
| DS Protein Docking | Predice las interacciones proteína-proteína de nuevas dianas de forma rápida y precisa |
Herramientas para análisis de secuencias
| HERRAMIENTA | DESCRIPCIÓN |
| DS Sequence Analysis | Identifica homólogos para su secuencia de proteína buscando en las bases de datos instaladas localmente o disponibles en el sitio web del NCBI |
Herramientas para construcción y análisis de biopolímeros
| HERRAMIENTA | DESCRIPCIÓN |
| DS Biopolymer | Construir y modificar modelos de ácidos nucleicos (DNA, RNA), proteínas y péptidos, así como protocolos para calcular potenciales electrostáticos y energías de solvatación |
Herramientas de simulación
| HERRAMIENTA | DESCRIPCIÓN |
| DS CHARMm | Simulaciones de dinámica molecular y minimizaciones de energía energía en moléculas desde pequeños ligandos hasta complejos fisiológicos multicomponentes |
| DS CHARMm Lite | Realiza minimizaciones de ligando in situ usando la maquinaria de simulación de CHARMm |
| CFF Advance Class II Forcefield | CFF, de Consistent Forcefield, optimiza modelos de DNA, RNA, carbohidratos, lípidos, proteínas, péptidos y pequeñas moléculas con gran fiabilidad en la precisión de los resultados |
| Merck Molecular Force Field (MMFF) | Campo de fuerzas extensamente parametrizado para sistemas orgánicos y bio-orgánicos y para interacciones intermoleculares cruciales para la unión a enzimas |
| DS Analysis | Análisis y visualización de trayectorias de dinámicas moleculares obtenidas en experimentos de simulación con CHARMm sobre proteínas o complejos proteína/ligando |
Herramientas para interacciones receptor-ligando
| HERRAMIENTA | DESCRIPCIÓN |
| DS Flexible Docking | Aproximación realista al docking flexible donde el docking de pequeñas moléculas se ve influído por conformaciones de baja energía de cadenas laterales en el sitio activo |
| DS LigandFit | Interacción de ligandos en un sitio de unión en un receptor macromolecular diana |
| DS LigandDdock | Docking eficiente usando características polares y apolores (hotspots) en el sitio del receptor para guiar el docking |
| DS LigandFit/CAP | Librería de ligandos en representación 3D obtenidos a partir de CAP (Chemicals Available for Purchase) y la base de datos CAPScreening |
| DS LigandScore | Evaluación objetiva de las interacciones ligando-proteína con funciones de puntuación y otros descriptores individuales |
| DS Ludi | Búsqueda de novo de fragmentos basado en los sitios de interacción en el bolsillo de unión al receptor para buscar librerías de fragmentos e identificar y clasificar moléculas |
| DS De Novo Evolution | Genera moléculas completas análogos de fármacos enlazando y añadiendo fragmentos sobre una estructura andamio |
| DS Ludi/CAP | Librería de fragmentos derivados de la base de datos CAP diseñada específicamente para Ludi; esta librería puede usarse para diseño de novo en modo receptor usando la maquinaria de Ludi |
Herramientas para modelado y análisis de farmacóforos
| HERRAMIENTA | DESCRIPCIÓN |
| DS Catalyst Build y DS Catalyst Search | Cree de forma sencilla bases de datos 3D para consultar modelos de farmacóforo e identificar potenciales compuestos cabeza de serie |
| DS Catalyst Hypothesis | Cree automáticamente modelos cuantitativos y cualitativos de farmacóforos que identifican las características químicas y estructurales esenciales necesarias para la unión diana |
| DS Catalyst Shape | Expanda o refine sus búsquedas desarrollando una representación 3D de una molécula a partir de una conformación específica para encontrar compuestos que satisfacen las restricciones espaciales 3D impuestas por el sitio de unión en el receptor |
| DS Catalyst Score | Evalúe y priorice rápidamente los compuestos de sus experimentos y consultas a las bases de datos |
| DS Catalyst Conformation | Calcule con rapidez modelos conformacionales para pequeñas moléculas ofreciendo diversidad de representaciones de las conformaciones de la molécula accesibles energéticamente |
| DS Catalyst Structure Based Pharmacophore (SBP) | Introducción rápida de patrones de actividad para entender los efectos laterales en los inicios del proceso de diseño de fármacos, con acceso a miles de modelos de farmacóforos y creación de nuevos modelos |
| DS De Novo Ligand Builder | Herramienta única de diseño basada en fragmentos que usa farmacóforos para guiar la colocación de los fragmentos |
Herramientas para QSAR y diseño de librerías
| HERRAMIENTA | DESCRIPCIÓN |
| DS QSAR y DS QSAR+ | Relaciones cuantitativas estructura-actividad, DS QSAR ofrece cientos de descriptores moleculares probados en sistemas biológicos para correlacionar con la actividad |
| DS Library Design | Completa suite de métodos de agrupamiento de similitud y diversidad desarrollados específicamente para el diseño de librerías químicas |
Herramientas para toxicología y ADMET
| HERRAMIENTA | DESCRIPCIÓN |
| DS ADMET Descriptors | Predicción de propiedades de absorción, distribución, metabolismo y toxicidad (ADMET) para colecciones de moléculas como candidatos de síntesis, librerías comerciales y colecciones de screening |
| DS TOPKAT | Emplea modelos robustos y validados QSTR (relaciones cuantitativas estructura-toxicidad) para evaluar la toxicidad y efectos medioambientales de los compuestos químicos |
 BIOVIA Materials Studio
BIOVIA Materials Studio